|
�P�D�͂��߂�
�@���i��������ɂ�����ł��d�v�Ȏ��g�݂�1������������ł��邱�Ƃ́C�N�����F�߂�Ƃ���ł��낤�B���̎��g�݂̒��ł��C�����㑕�u�Ɏc��c����������������v���Z�X�́C�c���������ׂď������邱�Ƃ�����i�����ł͂Ȃ����C�R�X�g�E���Ԃ����Y�R�X�g��肩����Ƃ�������j�Ȃ��Ƃ���C�����������X�N�������C�ߋ��ɑ����̐��i������������Ă���B����C�K�����ǂ����ɂ����Ďc���������o���E�ȉ��Ƃ��邱�Ƃ����߂Ă��Ȃ�1�j�B�����������Ƃ���C�ǂ��܂Ő���̎c������F�߂�̂��C�܂��c���l����Ɉ��̊�l�ȉ��ƂȂ���v���Z�X���ǂ�����Ċm������̂��C���o���f�[�V�����͊�ƂɂƂ��Ĕ��ɑ傫�ȉۑ�ƂȂ��Ă���B�{�e�ł́C����̎c�����x�l�ݒ�ƋߔN���i�J���̒��S�ɂȂ����o�C�I���i�������C���̐��o���f�[�V�����ɏœ_�������ďЉ��ƂƂ��ɁC���C�t�T�C�N����ʂ������o���f�[�V�����ɂǂ����g�ނׂ����ɂ��āC���҂�̍l�����Љ��B�Ȃ��C���o���f�[�V�����ɂ��ẮC�������̉ۑ肪����C��������ׂĂ��̎��ʂŏЉ�邱�Ƃ͕s�\�ł���B�֘A����Q�l�����������o�ł���Ă���̂ŁC�����̂�����͂�����������2-8�j���Q�ƒ��������B
�Q�D���o���f�[�V�����ɂ�����c�����x�l�ݒ�̍l����
�@�����ł͐���̎c�����x�l�Ɋւ��ė��j�I�Ȍo�߂����Ă������C�傫�ȓ]�@��1��1993�NEli
Lilly�̌����҂����\�����_���i�}1�j9�j��FDA�����o���f�[�V�����Ɋւ��鍸�@�������̃K�C�h10�j���쐬�������Ƃɂ���ƒ��҂͍l���Ă���B

�}1�@Fourman��ɂ��ŏ��̘_��
2.1�@���j�Ɍ������̎c�����x�l
�@1963�N�C�ŏ���cGMP���č��œ������ꂽ���C���̎��K�����ǂ����߂Ă����̂́C�g���u���ڎ��ŃN���[��
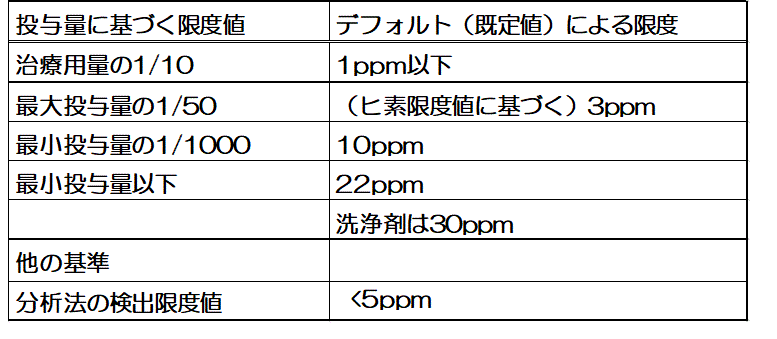
�iVisually Clean�j�h�Ƃ������Ƃł������B�������C���̔��f�ɂ͉Ȋw�I�ȍ��������o�����Ƃ͍���ł���C�����͊e�Ђ����ꂼ��̍l�����Ɋ�Â��Đ���̌��x�l��ݒ肵�Ă����B���Ƃ��C1992�N��PhRMA���s�����A���P�[�g����11�j�ł́C�\1�Ɏ������悤�Ȍ��ʂƂȂ��Ă���B�\�����ĕ�����悤�ɁC�e�Њe�l�ł���C�����ɂ͌�q����Eli
Lilly�̘_���ɂ݂���悤�Ȕ��f����܂܂�Ă���B�܂��C1993�NBarr Laboratory��FDA�̍ٔ�����12�j���o�āC�K�����ǂɂ��鐻������̍��@�������}���邱�ƂɂȂ�C���R�̂��ƂȂ�����E���o���f�[�V�����ɑ��Ă��C��������Ƃ������g�݂���Ƃɋ��߂��邱�ƂɂȂ����B�����āCFDA�͐��o���f�[�V�����Ɋւ��鍸�@�����̃K�C�h�gGuide
to Inspections�F Validation of Cleaning Processes�h10�j���쐬���C���̒��Ŏc�����x�l�ɂ��āC���̂悤�ɋL�ڂ��Ă���B
FDA ���@�������K�C�h10�j�ɂ�����L�ځF
�@FDA �́C���v���Z�X���o���f�[�g����Ă��邩�ǂ����f���邽�߂̋��e��₻�̕��@��ݒ肷�����͂���܂���B����эŏI���ܐ����Ŏg�p�����@���i�ɂ͑傫�Ȃ�������邽�߁CFDA
������̊�����߂邱�Ƃ͌����I�ł͂���܂���B�c�������x�l�̐ݒ荪���́C�����Ǝ҂��L����֘A�m���Ɋ�Â����_���I���̂ł���C�܂����p�I�C�B���\�C���؉\�Ȃ��̂ł���K�v������܂��B�����I�Ȍ��x�l��ݒ肷��ɂ́C���͕��@�̊��x���l�����邱�Ƃ��d�v�ł��B������v���[���e�[�V�����ŋƊE�̑�\�҂����y�������x�l�ɂ́C10
ppm �Ȃǂ̕��͌��o���x���C�ʏ�̎��×p�ʂ� 1/1000 �Ȃǂ̐����������x���C�ڂɌ�����c�������Ȃ��Ȃǂ̊��\�I�Ȋ�Ȃǂ�����܂��B
�@���x�l��ݒ肷��ꍇ�C���̕��@���m�F���邱�ƁB���w�I�������킩���Ă���c�����i�܂�C���������C�s���������C��܂Ȃǁj
�Ƃ͈قȂ�C�����v���Z�X�ɂ́C���w�I�ȓ��������m�ł͂Ȃ������I�Ȕ�������s�v�ȕ��Y�������݂���\��������܂��B�c�����E�l��ݒ肷��ꍇ�C���̉��w�ω���r���o���Ȃ��\��������C��v�Ȕ����������ɏœ_�Ă邾���ł͕s�\���ȏꍇ������܂��B�܂��C���w���͂ɉ����āCTLC
�X�N���[�j���O���K�v�ɂȂ�ꍇ������܂��B���ɃX�e���C�h�Ȃǂ̔��ɋ��͂ȉ��w�����̏ꍇ�C�����H���ɂ����āC��p�̑��u���Ȃ����ɂ͕��Y���̖����l������K�v������܂��B�o���f�[�V�����̖ړI�́C���x�l�̍������Ȋw�I�ɑÓ��Ȃ��̂ł��邱�Ƃ��m�F���邱�Ƃł��B
�\1�@1992�N��PhRMA�����{�����A���P�[�g��������
�@���̓��e�Ɠ��l�̋L�ڂ�1998�NHUMAN
DRUG CGMP NOTES13�j�ɂ��L�ڂ���Ă���C��Ƃ͎������������Y���鐻�i�̓������ӂ܂��āC�c�����x�l�̐ݒ���s�����Ƃ����߂��Ă���B�����āC���̍l�����͍����ł��ς���Ă��Ȃ��B
1998�NHUMAN DRUG CGMP NOTES13�j�ɂ�����L��
����F FDA�́C���o���f�[�V�����Ƃ���ɑ������m�F�ɑ��C�s�����Ɋւ��鋖�e���x�������Ă���̂��B
�F FDA�́C�����ٕ���������������̖��ɏd��ȊS�������Ă��܂��B�������������ɂ́C�O�̐��i��c�����n�t�̒P�Ȃ�L�����[�I�[�o�[�����ł͂Ȃ��C���܂�E�ʊ����܂̃L�����[�I�[�o�[���܂܂�܂��B�y�j�V�����������C
FDA�͐��o���f�[�V�����ɑ���W���I�ȋ��e���x�l���m�����Ă��܂���B���u�������鐻�i�̕����L�����Ƃ���C���ǂ�����̊�����߂邱�Ƃ͌����I�ł͂���܂���B�������CCGMP�̗v�������Ƃ́C���ِ���L���镪�͖@�Ɠ��l�ɐ��肷��K�v������܂��B�������̊�Ƃ́CICH�s�����K�C�h���C����U.S.P.General
Note �̗����ŋc�_����Ă���s��������臒l�Ƃ��Ă�0.1�������e���x�Ƃ��Č���ēK�p������Ƃ�����܂��B
����0.1���s��������臒l�̓K�p�͕s�K�Ƃ����܂��B�Ȃ��Ȃ���̌��x�l�͐����v���Z�X�ɊW�����s�����C���͊֘A�����̕]�����Ӑ}�������̂ł���C���������ɂ��O���ٕ��ɑ�����̂ł͂Ȃ�����ł��B���e���x�l�́C���v���Z�X�̔\�͂f���Ă��邱�Ƃ��d�v�ł���C���e���x�����߂鎞�ɂ́C�ȉ��̂悤�ȗv�����܂߂�ׂ��ł��B
�@�@�@�i1�j �L�����[�I�[�o�[�̗Տ���̉e���]��
�@�@�@�i2�j ���������̓Ő�
�@�@�@�i3�j �����X�t���̉��������̔Z�x
�@�@�@�i4�j ���͖@�̌��o���E
�@�@�@�i5�j �ڎ������@
�@�@�@ �����������q�̌������Ă��܂����C�ڎ������݂̂Ɉˑ����邱�Ƃ͉Ȋw�I�ɓK�Ƃ͂����܂���B
�@���̂悤�ɁC�K�����ǂ��ꗥ�Ɏc�����x�l�����̂悤�ɐݒ肵�Ȃ����ƌ������ƂL���������͂Ȃ��ƍl����ׂ��ł���B�ł́C�ǂ̂悤�ȕ��@�����邩�ł��邪�C�܂���Fourman��̍l����9�j���Љ��B
2.2�@Fourman��̊�Ɋ�Â����x�l�ݒ�>9�j
�@1993�NEli Lilly��Fourman��́C����3�̊�̒��ōł�����������̗p���ׂ����ƒ�Ă����B
�i1�j 0.1����F����ɐ��Y���鐻�i��1���ő哊�^�ʂɑ��āC�O���i�����̍ŏ����^�ʂ�0.1���ȉ��Ƃ���B
�i�����j0.001�̒��ɂ́C3��10 ���܂܂�Ă���B�ŏ���10�́C���i�͈�ʂɏ�������铊�^�ʂ�0.1
�Ŋ������Ȃ��Ȃ�ƍl�����邱�Ƃł���B2�Ԗڂ�10 �́C���S�W���ł���B������3�Ԗڂ�10�́C���o���f�[�V�������挒����L������̂ł���C�܂�C���ꂪ�\���������K�i�Ƃ��Đ��E�Ŏ�����邽�߂̂��̂ł��B
�i2�j 10ppm��F�O�ɐ����������i�̐��������ɐ������鐻�i�ɍ���������x��10ppm�Ƃ���
�B
�i�����j�ő勖�e100������1���x�����g�p����Ƃ����l���́C�H�i�ɓK�p�����K���i���F�_���̒��ł͋�̓I�Ȋ�͎�����Ă��Ȃ��j�ɂ��̃��[�c������܂��B���������K���ł́C�H���A���̒��ł�����ʂ̗L�Q�����������g�D��Ƌא��i�ɓ����Ă��Ă��������ƍl���Ă��܂��B
�i3�j �ڎ���F�ڎ��Ŋm�F���āC�c������F�߂Ȃ��B
�i�����j�����̏ꍇ�C����ɑ��u�ɖڎ��\�Ȏc�������c���Ă��Ă��C�ŏ���2�̊�ɓK�����Ă��܂��i���Ƃ��C�����i�g���E����USP���j�B���Ƃ��C�N���[���ƕ\������Ă��Ă��CGMP�̑��u�Ɏc�������ڎ��Ŋm�F�ł���͓K�Ƃ͂����܂���B����䂦�C�����C���S�Ȏc�����̗ʂ��ڎ��Ŋm�F�\�Ȃ�C���̏ꍇ���u�͎c�����������Ȃ��Ȃ�܂ŃN���[���ɂ��Ȃ��Ă͂Ȃ�܂���B
�@���̍l�����́C�����ł��L��������Ă��܂��B�Ȃ��C10ppm�͌��\��������ł��邱�Ƃ��C���ۂɌv�Z���Ă݂�ƕ�����B����C����3�̊�ł��邪�C���ۂ̌v�Z���i�{�e�ł͎����Ă��Ȃ��̂ŁC����2,3�j���Q�Ƃ��������j�ɂ͕s�m��W���Ȃǂ��܂܂�Ă���C�@�ݒ�̔w��ɉȊw�I�ȍ������Ȃ��C�A���������ŕs���v�������҂̎��_����̊�ƂȂ��Ă��Ȃ��C�Ƃ̎w�E������C�gWhy
the 10-ppm Criterion should be Abandoned�h�̂悤�ȃ^�C�g���̘_��14�j�����\����Ă���B�����������Ƃ���C����2015�N�ȍ~�c�����̓Ő��Ɋ�Â������x�l�ݒ�̍l�������L�����y���Ă���C����Fourman��̊�Őݒ肵�Ă����Ƃ��Ă��C�Ő��̎��_�ł̎c�����x�l���������Ă��Ȃ��ƍ��@�ŕK��������邱�ƂɂȂ�B
2.3�@�c�����̓Ő��Ɋ�Â����x�l�ݒ�
2.3.1�@����GMP�K�C�h���C��15�j
�@2001�NICH Q7 ����GMP�K�C�h���C�����ʒm���ꂽ���C�g12.7
���̃o���f�[�V�����h�ŁC���̂悤�ɋL�ڂ���Ă���B12.74 �c�������͉����������o�ł��銴�x��L����o���f�[�V�����ς݂̕��͕��@���g�p���邱�ƁB�e���͕��@�̌��o���E�́C�c�������͉������̋��e���������o����̂ɏ\���Ȋ��x�Ƃ��邱�ƁB���Y���͕��@�̒B���\�ȉ��������ݒ肷�邱�ƁB
�c�������E�l�́C���p�I�C�B���\�C���؉\�ł���C���C�ł��L�łȎc�����Ɋ�Â������̂Ƃ��邱�ƁB���E�l�́C���͂��̍ł��L�łȑg�����Ɋւ�����m�̖w�I�C�Ő��w�I���͐����w�I�����̍ŏ��ʂɊ�Â��Đݒ肷�邱�ƁB�i�����C�^���b�N�͒��ҁj
�@���̂悤�ɁC�������C���ł�2001�N�̒i�K�ŁC�c�����̓Ő��ɂ��ƂÂ������x�l�̐ݒ肪�w�E����Ă���B�������C�ǂ̂悤�ȓŐ��f�[�^���g�p���邩���L����Ă��炸�C�܂����҂�2015�N�̒i�K�ŕ����̌������[�J�Ɋm�F�����Ƃ���C�K�������Ő��Ɋ�Â��ݒ�����Ă���Ƃ͓̉���ꂸ�C�C�^���b�N�̕����Ȃǂ������ɐݒ肵�Ă������̂Ɛ��@���Ă���B�Ȃ��C���ڐ��Ƃ͊W���Ȃ����CICH
Q3C �g���i�̎c���L�@�n�}�K�C�h���C���h�i1998�N�j�̒��ɂ́C�Ő��Ɋ�Â��c�����x�l���v�Z���鎮��������Ă���C�Ő��f�[�^�Ƃ���PDE�iPermitted
Daily Exposure�j���g�p����Ă����C����PDE���C���̌����̎c�����]���̕\����ɏo�Ă��邱�ƂɂȂ�B
2.3.2�@EU-GMP�K�C�h���C��16�j
�@2014�N���B���i���iEuropean Medicines
Agency�FEMA�j�́C��Ǝ҂̈��S���Ɏ��_��u�������X�N�Ɋ�Â��\�I�Ǘ��Ɋւ���K�C�h���C��
�gGuideline on setting health based exposure limits
for use in risk identification in the manufacture
of different medical products in shared facilities�h��ʒm���C2015�N����{�s�ƂȂ����B���̃K�C�h���C���͐��o���f�[�V������ΏۂƂ������̂ł͂Ȃ����C�ȉ��̂悤�ȋL�ڂ�����C���̌��ʁC���̃K�C�h���C���������ɓŐ��Ɋ�Â�����̎c�����]�������y���邱�ƂƂȂ����B
�@�i�G�O�[�v�e�B�u�T�}���[�j�������������̑��݂́C�S�Ă̐l�X�ɂƂ��Ĉ��S�ƍl�����郌�x���̏��Ɏ�����郊�X�N�Ɋ�Â��ĊǗ�����Ȃ���Ȃ�܂���B���̂��߁C���S臒l���v�Z���邱�ƂŌ��N�Ɋ�Â������x�l�����X�N����肷�邽�߂ɍ̗p����Ȃ���Ȃ�܂���B��������臒l�̌v�Z�i�Ⴆ�C1���ő�ێ拖�e�ʁiPDE�j�C���邢�́@�Ő��w�I���O��臒l�iTTC�j�j�́C��Տ��C�Տ������f�[�^���܂ޗ��p�ł��邷�ׂĂ̖w�I�C�Ő��w�I�ȃf�[�^�𑍍������Ȋw�I�ȕ]���łȂ���Ȃ�܂���B
�@�iIntroduction�i�����j�j���̓��X�N�ጸ��ł���C����ƊE�ł͐��o���f�[�V�����̃L�����[�I�[�o�[���x�l���L���g�p����Ă��܂��B�����̌��x�l���m�����邽�߂ɂ��܂��܂ȃA�v���[�`������Ă��܂����C�����̏ꍇ�C���p�\�Ȗw�I����ѓŐ��w�I�f�[�^���l������Ă��܂���B���������āC���X�N�̓���Ƃ��ׂẴN���X�̈��i�����̃��X�N�ጸ����T�|�[�g����ɂ́C���Ȋw�I�ȃP�[�X�o�C�P�[�X�̃A�v���[�`���K�v�ł��B���̃K�C�h���C���̖ړI�́C�X�̗L�������̖w�I����ѓŐ��w�I�f�[�^�����r���[����ѕ]�����CGMP�K�C�h���C���Ō��y����Ă���臒l���x��������ł���悤�ɂ���A�v���[�`�𐄏����邱�Ƃł��B�����̃��x���̓��X�N����c�[���Ƃ��Ďg�p�ł��C���o���f�[�V�����Ŏg�p�����L�����[�I�[�o�[���x�𐳓������邽�߂ɂ��g�p�ł��܂��BGMP�K�C�h���C���̑�3�͂Ƒ�5�͂ł͗L�����i�����iAPI�j�ɂ��Ă͐�������Ă��܂��C���X�N����̂��߂�臒l���o�����߂ɂ��̃K�C�h���C���ŊT������Ă����ʌ����́C�K�v�ɉ����ēK�p�ł��܂��B
�@���̂悤�ɁC����̎c�����x�l��Ő��Ɋ�Â��Đݒ肷��ꍇ�C���̃K�C�h���C�����q�ׂĂ���PDE��TTC���g�p�ł��邱�Ƃ����m�Ɏ�������Ă���B2014�N�H���҂�͉��B�̍��@�������C���̎����łɓŐ��Ɋ�Â����x�l�]�������Ă��Ȃ��̂��Ƃ̎�������B�������C���̎��̓K�C�h���C���̎{�s��2015�N�Ƃ������ƂŁg�������h�Ɖ��ė������B�������āC�����ł͓Ő��Ɋ�Â��c�����x�l�̕]���i���̐��l��ݒ�邩�ǂ����͕ʂƂ��āj�͕K�{�̗v���ƂȂ��Ă���Ɨ������ׂ��ł���B�Ȃ��C���̃K�C�h���C���������Ă���PDE�̌v�Z����}2�Ɏ����ƂƂ��ɁC���ۂɃL�����[�I�[�o�[���v�Z���鎞�̎��������Ď������B�����番����悤��Fourman��̌v�Z���ɂ�����10ppm��0.1���ɑ������镔����PDE�̐��l�����鎖�Ō��x�l��ݒ肷�邱�ƂɂȂ�B�Ȃ��C���Ⴂ���Ȃ������悢�Ǝv���̂́C�K�C�h���C���͓Ő��Ɋ�Â����x�l��ݒ肵��ƌ����Ă���̂ł͂Ȃ��i�g2.5�@���x�l�Ԃ̔�r�h���Q�Ɓj

�}2�@PDE�y��Carry-Over�̌v�Z��
�@
2.4�@�H���\�͂Ɋ�Â����x�l�ݒ�
�@�c�����x�l�̐ݒ���@�Ƃ��āC����1�̍l�������Љ�Ă����B����́C���炩���߃f�t�H���g�l�Ƃ��Đݒ肳��Ă���C���Ƃ���WFI�ɂ�����TOC�l�Ȃǂ𗘗p������C���邢�͂�Fourman��̎菇�Ō��x�l��ݒ肵�Ă����C���̌�͎��ۂɐ���Ɏc�����Ă���ʂ���Ɍ��x�l������������Őݒ肷��Ƃ������̂ł���B���̊�ł���C���Ƀv���Z�X�ɖ�肪�������ꍇ�C�ʏ�ƈقȂ�Ƃ̔��f���o���邱�ƂɂȂ�B�����Validation�{���̖ړI�ƍ��v���Ă���̂ł͂Ȃ����ƒ��҂͍l���Ă���B���̂��߁C�ŏ��̒i�K�͓Ő���Fourman��̌v�Z���Ɋ�Â����x�l�Ƃ��Ă����C���ۂɐ��Y���J�n��������l���W�ς��ꂽ�i�K�ŁC���������H���\�͂Ɋ�Â����x�l�ɕύX���ׂ��ł͂Ȃ����Ȃ����낤���B�{���ł͏q�ׂȂ����C���̍l������Quality
by Design�Ɋ�Â����o���f�[�V�����̎��g�݂ƈ�v������̂ƍl���Ă���B
2.5�@���x�l�Ԃ̔�r
�@����܂ŏЉ�Ă����悤�ɁC�c�����x�l�̌v�Z�菇�́C���낢��ȕ��@������BBarle��17�j�́C140�̌���ɂ��āC�Ő��Ɋ�Â����x�l��Fourman���0.1����Ɋ�Â��v�Z�ł́C90���̖���҂̕����������l�ɂȂ����ƕ��Ă���i�}3�j�B���̑��ɂ��������C�������C���ɂ�������x�l�ɂ��ē��l�̕����Ă���18�j�B���̂悤�ɁC�����̖̏ꍇ�CFourman��̊�̕������x�l�Ƃ͌������Ȃ�X���ɂ���B�ł́C����Ƃ��āC��������m�œŐ��Ɋ�Â����x�l�����x�l�Ƃ��č̗p���邩�ł���B���҂��������Ă����g�D�ł́C�ŏI�I�ɇ@Fourman��̊�C�A�Ő��Ɋ�Â���C�̂ǂ��炩�������l���̗p���邱�ƂƂ����B
�@�Ȃ��CISPE�����s���Ă���Baseline guide19�j�ɂ́C���̂悤�ɋL�ڂ���Ă���B
�@���o���f�[�V�����ɂ�������x�l�̐ݒ�ɂ����āC�Œ�Տ��p�ʁiLCD�j��1/1000�C������10ppm�͏�ɓK�ȁi����ҁj�ی�Ƃ͂Ȃ�Ȃ��ꍇ������B���Ƃ��C�z��������R������ᇍ܂̂悤�ȃn�U�[�h���x���̍����������ɂ��Ăł���B
�@�������C�����̖ɂ����āC���̕��@�͌��N��Q�������҂���邽�߂̋��e�ł���c�����x�l�ƍl���邱�Ƃ��ł���B
�@�܂�C�Ő��Ɋ�Â����x�l�ݒ肪�d�v�Ȃ̂ł͂Ȃ��C���o���f�[�V�����{���̖ړI�₱�������R�����g�����܂��C���ЂŐ���̎c�����x�l�ݒ�ɑ��Ă�������Ƃ����l���������đΉ����邱�Ƃ��d�v�Ȃ̂ł���B
�@
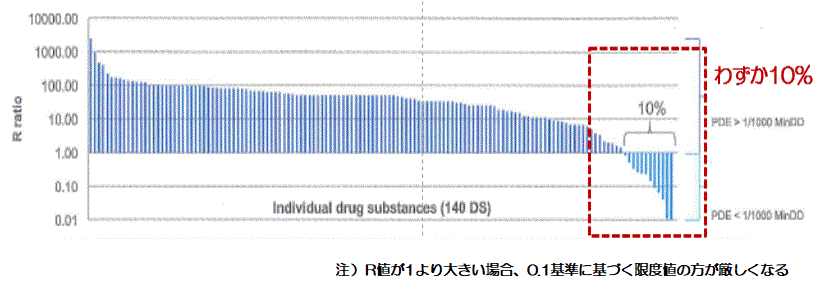
�}3�@PDE�Ɋ�Â����x�l��0.1����ɂ���Â����x�l�̔�r
�@�������́u����PHARMSTAGE�v�Q�O�Q�T�N�Q�����@�{���ł�������������
�@�@����PHARMSTAGE�̃z�[���y�[�W�͂�����
�@�@https://www.gijutu.co.jp/doc/magazine_pharm%20stage.htm
�Q�l����
1�jFDA, Human Drug CGMP
Notes, 9 : 2, 2Q2001
2�j�m���S�W�n�����ݔ��̐��o���f�[�V������3�ɗv�������Ή�
�@�T�C�G���X���e�N�m���W�[�i���j�C2013�N
3�j�{�����t�C�o���^���@�w�E�^���������E�K�����瓱�����E���o���f�[�V�����F���f��ƃm�E�n�E�C�T�C�G���X���e�N�m���W�[�i���j�C2021�N
4�jKenneth E. Avis (Ed.),
Biotechnology Quality Assurance and Validation,
CRC Press (2019)
5�jRoger Brunkow et al.,
Cleaning and Cleaning Validation: A Biotechnology
Perdpective, CRC Press (1996)
6�jDestin A. Blanc, Cleaning
Validation Practical Compliance Approaches for
Pharmaceutical Manufacturing, CRC Press (2023)
7�jISPE, Cleaning Validation
Lifecycle-Applications, Methods, Controls, ISPE
(2020)
8�jPDA, Technical Report
No. 49 : Points to Consider for Biotechnology
Cleaning Validation. (2010)
9�jFourman, G. L. and Mullen,
M. V., Determining Cleaning Validation Acceptance
Limits for Pharmaceutical Manufacturing Oprtations,
Pharm. Technol. 17 (4), 54-60 (1993)
10�jFDA, Guide to Inspections
: Validation of Cleaning Processes (1993)
11�jAndrew Walsh, Cleaning
Validation for the 21st Century : Acceptance Limits
for Active Pharmaceutical Ingredients : Part I,
PHARMACEUTICAL �@ENGINEERING, July/August, 74-83
(2011)
12�j���R�o�c�������CBarr Laboratories���������̂��ȁH
�@https://ncogmp.com/2023/03/09/post-13156/
13�jFDA, HUMAN DRUG CGMP
NOTES , Volume 6, Number 2, June, 1998
14�jAllan Ader, Cleaning
Limits-Why the 10-ppm Criterion should be Abandoned,
Pharm. Tech., 40 (1), 52-56 (2016)
15�j�����J���ȁC����GMP�̃K�C�h���C���ɂ��āC���1200��
�i2001�N11��2���j
16�jEMA, Guideline on setting
health based exposure limits for use in risk identification
in the manufacture of different medical products
in shared facilities�i2014�N11��20���j
17�jE.L.Barle, Using Health-Based
Exposure Limits to Assess Risk in Cleaning Validation,
Pharmaceutical Technology
�@https://alfresco-static-files.s3.amazonaws.com/alfresco_images/pharma/2017/09/19/3ffbe23f-bab2-4774-ad65-b94ed8ab270c/PT0817%20Lonza%209-6%20ES%20pr2f-Web.pdf
18�j���g���F�C���o���f�[�V�������{�E�T���v�����O�Ó�����DHT�ECHT/�c�����e�l�̐ݒ�C�T�C�G���X���e�N�m���W�[�i���j�Cpp.
29-48�i2017�j
19�jISPE Baseline guide
: Volume7_Risk-Based Manufacture Of Pharmaceutical�@Products
First Edition (2010)
|


