|
1.はじめに
医薬品の製造販売承認申請は,各地域の規制当局により審査され,製造販売承認が与えられる。承認後に,CMC
に関する変更を行う際には,各地域の規制に従って承認される必要がある。各地域で承認された製造販売承認に関するすべての変更の責任は製造販売業者(MAH)にあり,これらの変更のマネジメント及び維持管理を行う必要がある。
様々な世界の市場の患者に医薬品を届けるために,各地域の要求事項に対応する必要があり,同一の医薬品に対して各国向けの異なる製造方法や規格を設定することが必要になることがある。これらの場合には各国向け製品の在庫管理も必要となり,グローバルサプライチェーンによるCMO
の増加と相まってより複雑化している。また,商用生産段階では様々なCMC に関する変更が生じるが,その申請や審査についても各地域の規制が異なっていることから,一つの変更について,各地域で手続きを完了するまでの期間はおよそ4〜6
年間と長期化している。
ICH Q12 の協議が始まる前は,ICH の品質分野において医薬品の開発から承認までのライフサイクルの早期に焦点が当たり,医薬品ライフサイクルを補完する承認後の変更に関しての柔軟な運用は実現されておらず,承認後の変更に関して要求される資料や薬事手続きが調和されていないという課題が解消されず,変更によるイノベーションや継続的改善を阻害することにも繋がっていた。
これらの課題を解決するために,医薬品ライフサイクルを通した承認後のCMC 変更をマネジメントする枠組みを提供するICH
Q12「医薬品のライフサイクルマネジメントにおける技術上及び規制上の考え方に関するガイドライン」1)が作成された。
(中略)
3.ICH Q12 の概要
(中略)
3.1 CMC に関する承認後変更の分類
承認後のCMC に関する変更は,各国の規制に従って行われる必要がある。CMC
に関する変更には,製品品質に与える潜在的リスクが高いものから低いものまであり,リスクが高いものは企業と規制当局間のコミュニケーションが必要になる。各国の薬事手続きには,必要とされる情報及び必要な期間に違いがあるが,少なくとも「事前承認」と「届出」の規制の枠組みが必要となる。また,規制当局への報告が必要ない変更については,PQS
内で管理されるが,この記録については定期的な調査で確認されることになる。
変更カテゴリーの分類は,表2の通りであるが,ICHQ12 ガイドラインの実装に向けて各国で規制の枠組みの改正が行われている。
表2 変更カテゴリーの分類
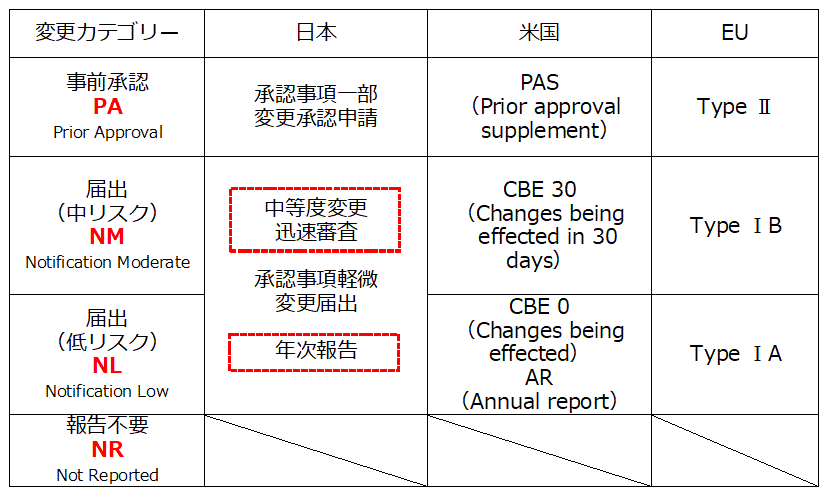
日本においては,「医薬薬審発0927 第4
号 中等度変更事項に係る変更手続の導入の試行について」2)により,中等度のリスクの変更について,中等度変更迅速審査という届出(中リスク)に相当する変更カテゴリーの導入が検討され,ICH
Q12 導入に伴うさらなる国際整合が図られようとしている。また,承認書の製造方法の記載方法等について軽微変更届出とされた事項について,品質に与える影響が比較的小さいものについて,年に1度の報告に代えることができる年次報告3)とすることができる制度を導入するために,試行的な取組が実施されている。なお,中等度のリスクの変更及び年次報告については試行段階であるため,本格実装の内容とは異なることがあるため注意が必要である。
承認後変更のリスクに基づく分類の仕組みの調和は,企業と規制当局間の透明性を高め,継続的改善につながることが期待される。
3.2 エスタブリッシュトコンディション(EC)
エスタブリッシュトコンディション(EC :
Established Condition)とは,製品品質を保証するために必要と考えられる法的拘束力のある情報と定義されており,ECの変更には薬事手続きが必要となる。日本では,医薬品の承認申請書における製造方法の記載方法等について,平成17
年2 月10 日に発出された旧通知「改正薬事法に基づく医薬品等の製造販売承認申請書記載事項に関する指針について」4)に従って記載してきたので,概念としては理解されやすい。医薬品の承認審査において,企業と規制当局が承認時に品質確保に必要な事項を合意し,承認後に承認事項を遵守するというコンセプトに基づいている。日本で先行していた承認申請書の概念はICH
Q12 でEC として広くICH 加盟地域において適用されることとなる。承認申請書の記載については,令和6年9
月30 日医薬薬審0930 第7 号「医薬品の製造方法の記載方法及び変更手続について」5)が示され基本的な考え方が整理された。
MAH が製造販売承認申請(MAA : Marketing Authorization Application)の際に,規制当局にデータ又は情報を提供するためのCMC
に関する薬事上のコミットメント(安定性,CMC に関する承認後のコミットメント及びその他のコミットメント等)についてはECとは区別されて管理され,ICH
Q12 では参考情報とみなしている。CMC に関する薬事上のコミットメントの変更については,各地域における現行の規制及びガイダンスに従い管理される。
(1)製造工程のEC の特定について
製造工程のEC は,製品及び製造工程の理解に基づき,管理戦略の関連する要素をすべて考慮して特定される。単位操作及びステップの順序に加え,全体の管理戦略を考慮し,妥当性を示して提案すべき製造工程のEC
は,製品品質を保証するために必要なインプット(工程パラメータ,物質特性等)及びアウトプット(工程内管理)となる。
要求される品質の製品が製造されることを保証するために管理を要する工程パラメータはEC とみなされ,これらのEC
はクリティカリティの評価により特定される。クリティカリティの評価では,危害の重大性やEC の変動範囲が適切に考慮されているかを確認する必要がある。重要工程パラメータ(CPP
: Critical Process Parameter)や製品品質に影響を及ぼす可能性が否定できないその他の工程パラメータは,EC
として設定すべきである。
EC を特定した後は,全体の管理戦略を考慮し,ICH Q9 に基づくリスクアセスメントを行い,EC
の変更カテゴリーを決定する。リスクアセスメントの結果により,EC の変更が高リスクから低リスクまで分類され,リスクレベルに応じた変更カテゴリーを判断する。EC
の変更の潜在的リスクと変更カテゴリーの根拠については明確に示す必要がある。
製造工程のEC を特定する手法には,パラメータに基づく手法と性能に基づく手法がある。パラメータに基づく手法には,ICH
Q8 及びICH Q11 で示されている通り,経験に基づいて限られた製造条件のもとで開発を行う最小限の手法(Minimal
Approach)と,クオリティ・バイ・デザイン(QbD)の原則を取り入れたより進んだ手法(Enhanced
approach)がある。さらに,性能に基づく手法(Performance-based Approach)として,より進んだ手法から得られた知識,豊富なデータが得られる環境及びより進んだ管理戦略(モデル,プロセス解析工学(PAT
: Process Analytical Technology) 等)によって製造工程の理解が進んだ環境では,工程のインプット(工程パラメータ,物質特性等)よりも,工程のアウトプット(特性,測定値,反応等)の管理に重点を置いた管理戦略を使用することも可能となる。
(2)分析法のEC の特定について
分析法に関するEC には,分析法の性能を保証する要素を含めるべきである。ICH
Q146)で今後示されるツールを用いて,分析法の性能を保証するために管理が必要なパラメータ等を特定することができる。EC
の範囲及びそれらの変更カテゴリーは,分析法のパラメータと性能の関係に関する理解度,分析法の複雑さ及び管理戦略により異なる。
分析法の開発手法についても,開発段階で実施された検討が限定的である従来の開発手法に加えて,より理解が深まっている場合には,適切なEC
を提案することによって,分析法の性能を確保するための管理幅が広がったり,それによってEC を減らし分析法の性能に重点を置いたものにしたりすること(分析法パラメータの設定値ではなく許容範囲や性能基準をEC
とする等)ができるようになる。
3.3 承認後変更管理実施計画(PACMP)
承認後変更管理実施計画は,承認を受けた品目について承認された事項の一部の変更に係る変更計画(PACMP:
Post Approval Change Management Protocol)であり,ICH
Q12 にて考え方が示され,令和3年8月1日より本格導入された変更制度である。医薬品等の製造販売承認後の品質に係る承認事項の変更に係る予測性及び透明性の向上に資するよう,製造販売業者等とPMDA
とがあらかじめ,製造方法等の変更内容,変更内容に対する評価方法及び判定基準,品質に係る承認事項の変更案,医薬品等適合性確認の要否等について合意しておき,その後,合意された評価方法に従って検討を行い,予定された結果が得られた場合は,届出により,品質に係る承認事項を予定していた案へ迅速に変更できる制度である。本制度については,米国ではComparabilityProtocols
として,欧州ではPost-Approval ChangeManagement Protocols(PACMPs)として制度化されていたものであるが,日本においても,ICH
Q12 の実装により国際整合するため,「変更計画」7)として実現した。PACMP
は,変更の実施に必要な要件と検討に関してMAH と規制当局の間で事前に合意するので,変更の予見性が向上する。
3.4 製品ライフサイクルマネジメント(PLCM) の文書
製品ライフサイクルマネジメント(PLCM :
ProductLifecycle Management)の文書は,MAH がCMC に関する承認後変更のマネジメントをどのように行う計画であるかを規制当局に明確に伝える要約文書である。PLCM
の文書には,EC,EC 変更時の変更カテゴリー,PACMP,及び承認後のCMC に関するコミットメントが含まれる。PLCM
の文書については,初回の製造販売承認申請(MAA)時または市販品のEC を特定する薬事手続きを行い際に提出する必要がある。また,CMC
に関する承認後変更の薬事手続き時には,更新したPLCMの文書を含める必要がある。
PLCM の文書の具体例(表3)については,ICH Q12ガイドラインの付属書IF に示されている。PLCM
の要素の内,EC 及びその変更カテゴリーについては表形式での記載が推奨されているが,各地域の規制に従って記載する必要がある。
PLCM の文書は,日本においては承認申請書を運用しているため理解されやすい概念である。PLCM
の文書の要素の内,EC 及びそれに関連する変更カテゴリー,承認後のCMC に関するコミットメントは承認申請書で運用される。また,PACMP
は,PACMP 制度である「変更計画」で運用されることになる。これらのPLCMの文書の記載箇所については,今後の規制の動向やICH
M4Q(R2)8)を参照されたい。
表3 PLCM の文書の具体例

(中略)
4.製品ライフサイクルの管理
製品ライフサイクルの早期段階(製品開発,製造販売承認申請及び上市)については,ICH
Q8(R2),Q9,Q10 及びQ11 に示され,商業生産段階の柔軟な薬事対応についてはICH
Q12 にその概念が示された。製薬企業では製品の開発段階に得られた製品理解に基づき,CMC
に関する承認後変更をPQS の変更マネジメントにより管理を行っている。承認後変更,特にEC
の変更を伴う変更は薬事手続きが必要であり,各地域の規制に従った変更の手続きが求められる。ICH
加盟地域におけるICH Q12 の実装状況についてはICH Q12 のWebページ9)を参照されたい。欧州(EU)は,「Note
on EU implementation of ICH Q12」10)において,EC
の特定とその変更カテゴリーに関する科学及びリスクに基づくアプローチ,及びPLCM の文書が,既存のEU
の薬事手続き(Variations)に関する法的枠組みには適合しないと考えられているので,現行の法規制に基づいた手続きが求められることに注意が必要である。
MAA を行った製品について,承認後の規制当局との共通認識を保つための文書として,各国のEC,その変更カテゴリー,PACMP
及び承認後のCMC に関するコミットメントを記載したPLCM の文書で管理が行われる。今後,さらに多くの地域でPLCM
の文書の実装が進むことにより,現行の法規制に従った各地域でのEC等が設定され,管理する内容がより明確となり,企業側と規制当局の間で,承認後変更の管理がより効率的に行われる。
ICH Q12 の実装は,製剤,原薬及び分析の開発から商業生産段階における管理文書の概念の調和が進むことにより,承認後の管理についても透明性が増し,規制当局の手続きの効率化が進み,また,これらのことによりMAH
による適切な管理がより促進され,グローバル市場への高品質な医薬品の安定供給が図られることが期待される。
◆本稿の全容は「月刊PHARMSTAGE」2025年4月号 本誌でご覧ください◆
月刊PHARMSTAGEのホームページはこちら
https://www.gijutu.co.jp/doc/magazine_pharm%20stage.htm
参考文献
1)ICH-Q12 医薬品のライフサイクルマネジメント
(PMDA web ページ:https://www.pmda.go.jp/int-activities/int-harmony/ich/0041.html)
2)令和6 年9 月27 日 医薬薬審発0927 第4 号 厚生労働省医薬局医薬品審査管理課長通知「中等度変更事項に係る変更手続の導入の試行について」
(厚生労働省 web ページ:https://www.mhlw.go.jp/hourei/doc/tsuchi/T240927I0010.pdf)
3)令和7 年2 月13 日 医薬薬審発0213 第5 号 厚生労働省医薬局医薬品審査管理課長通知「年次報告に係る変更手続導入に向けた試行的実施について」
(厚生労働省 web ページ:https://www.mhlw.go.jp/hourei/doc/tsuchi/T250214I0030.pdf)
4)平成17 年2 月10 日 薬食審査発第0210001 号厚生労働省医薬食品局審査管理課長通知「改正薬事法に基づく医薬品等の製造販売承認申請書記載事項に関する指針について」
5)令和6 年9 月30 日 医薬薬審0930 第7 号 厚生労働省医薬局医薬品審査管理課長通知「医薬品の製造方法の記載方法及び変更手続について」
(厚生労働省 web ページ:https://www.mhlw.go.jp/hourei/doc/tsuchi/T241001I0050.pdf)
6)ICH Q14(ステップ4)Analytical Procedure Development
(PMDAweb ページ:https://www.pmda.go.jp/files/000265999.pdf)
7)令和3 年6 月16 日 薬生薬審発0616 第14 号 厚生労働省医薬・生活衛生局医薬品審査管理課長通知「医薬品等の変更計画の確認申請等の取扱いについて」
(厚生労働省 web ページ:https://www.mhlw.go.jp/web/t_doc?dataId=00tc5989&dataType=1&pageNo=1)
8)ICH M4 : The Common Technical Document, M4Q(R2)
(ICH Webページ:https://www.ich.org/page/multidisciplinary-guidelines)
9)ICH Q12 : Technical and Regulatory Considerations
for Pharmaceutical Product Lifecycle Management
(ICH Web ページ:https://www.ich.org/page/quality-guidelines)
10)04 March 2020 EMA/CHMP/ICH/78332/20201
(EMA Web ページhttps://www.ema.europa.eu/en/documents/other/note-eu-implementation-ich-q12-guideline-technical-and-regulatory-considerations-pharmaceutical-product-lifecycle-management_en.pdf) |


