|
�P�D�͂��߂�
�@ICH Q8, Q9, Q10, Q11�y��Q12�K�C�h���C���ł́C�u�Ȋw�y�у��X�N�Ɋ�Â����i�J�����тɃ��X�N�Ɋ�Â��K�����ǂ̔��f�̋@�����Ă���B�v1�j
�����̃K�C�h���C���́C���i���C�t�T�C�N����ʂ��ĉ��w�C�����y�ѕi���Ǘ��iChemistry,
Manufacturing and controls, �ȉ� CMC�j�̕ύX�Ɋւ���]�����s����ŗL�p�ł���B
�@ICH Q8�y�� Q11�K�C�h���C���́C��ɐ��i���C�t�T�C�N���̏����i�K�i���Ȃ킿���i�J���C�����̔����F�\���y�т�ΏۂƂ��Ă���BICH
�K�C�h���C���̍ŋ߂̎��{�o������CICH Q8 (R2)�y��Q10�t���� 1�ɋL�q����Ă���
CMC�Ɋւ��鏳�F��ύX�ɑ���C���_��ȋK���̃A�v���[�`�����S�ɂ͎�������Ă��Ȃ��Z�p�ƋK���Ƃ̊Ԃ̃M���b�v�����炩�ɂȂ��Ă����B�{�e�ł́C���C�t�T�C�N���Ǘ��ɂ����鏳�F��ύX�Ǘ��iPACMP�j�Ƃ��̐i�ߕ��𒆐S�ɊT��������1-7�j�B
�@ICH Q12 �u���i�̃��C�t�T�C�N���}�l�W�����g�K�C�h���C���v�̎戵����ȓ��e�Ƃ��Ă͈ȉ��ł���B
�@���@ CMC�Ɋւ��鏳�F��ύX�F���̂ƋK�����ǂ̃��X�N�R�~���j�P�[�V����
�@���@ �G�X�^�u���b�V���g�R���f�B�V�����iEC�j�F���i�i�����m�ۂ��邽�߂ɕK�v�Ȏ�����C�ύX�ɍۂ��Ė葱�����K�v�Ȏ���
�@���@ ���F��ύX�Ǘ����{�v�揑�iPACMP�j�F���̂ƋK�����ǂ̎��O���ӂɂ��ύX�葱���̂��߂̃c�[��
�ł���B
�@���@ ���i���C�t�T�C�N���}�l�W�����g�iPLCM�j�F���F���CMC�Ɋ֘A�����R�~�b�g�����g�CPACMP�C���ƒi�K�ł̃}�l�W�����g���L��
�@���@���i�i���V�X�e�� �iPQS�j �y�ѕύX�}�l�W�����g
�@���@�K�����ǂɂ��R���ƒ����̘A�g
�@���@ �s�̐��i�̏��F��ύX�F���͖@����萫�����Ɋւ��ύX�𑣐i����A�v���[�`
�@�{�e�ł́CICH Q12�̒��ł��d�v�ȃR���Z�v�g�ł���EC�y��PACMP�ɏœ_�āC�ύX�����^�p�ʂ������ĊT������B
�Q�DICH Q12�ɂ�����K���̃c�[���ƒB���̂��߂̎�@
�@�G�X�^�u���b�V���g�R���f�B�V�����iEC�j�F
�@���F�\���ɍۂ��ẴR�����E�e�N�j�J���E�h�L�������g�iCTD�j�l���͌��߂��Ă��邪�C���F�\���ɂ����Đ��i�i�����m�ۂ��邽�߂ɕK�v�ƍl�����C���F��̕ύX�ɍۂ��Ė葱�����K�v�ȗv�f���K�肷�钲�a�����A�v���[�`�͂���܂łɂ͂Ȃ������B�{�K�C�h���C���iICH
Q12�j�ł́C���̂悤�ȗv�f���u�����y�ъǗ��Ɋւ���G�X�^�u���b�V���g�R���f�B�V�����v�i�{�K�C�h���C���ł�
EC �Ƃ���j�ƒ�`���Ă���B
�@��EC�̒�`�y�ыK�����ǂւ̒�o�����ɂ����������
2.1�@EC�̒�`
�@EC�́C���i�i�����m�ۂ��邽�߂ɕK�v�ƍl������@�I�S���͂̂�����i�܂菳�F�����j�ł���B���������āCECs
�̂����Ȃ�ύX���葱����K�v�Ƃ���BECs�K�����ǂւ̒�o�����ɂ́CEC �ƎQ�l��܂܂��B�Q�l����
ECs�Ƃ݂͂Ȃ���Ȃ����C�K�����ǂɑ��ĊJ���y�ѐ����Ɋւ��ēK�x�ɏڍׂȏ�����CEC�y�т��̕ύX�J�e�S���[�̑I���ɂ��č������������̂ł���B
�@CMC �Ɋւ���R�~�b�g�����g�̕ύX�ɂ��ẮC�{�K�C�h���C���ł͈���Ȃ����C�e�n��ɂ����錻�s�̋K���y�уK�C�_���X�ɏ]�����ƂɂȂ�B
2.2�@EC�̓���ƕύX�Ǘ��̎��{�v��
�@���̍��ł́C�����H���y�ѕ��͖@�� EC�����߂邽�߂̃A�v���[�`�̊T�v���L�q����B����
EC�i�e��{���n�̐��\���j�����߂�ۂɂ����l�̃A�v���[�`���g�p���邱�Ƃ��ł��邪�C�\���҂͂��̍�����������C�K�����ǂɔF�߂��Ȃ���Ȃ�Ȃ��BEC
�͈̔͂́C��Ƃɂ��J����@�y�ѐ��i�i���ɑ�����ݓI���X�N�ɂ��قȂ�B
�������H���� EC�̓��聄
�@�P�ʑ���y�уX�e�b�v�̏��ɕύX�J�e�S���[�̓�������肷��i�}1�j2,3�j�B
�@
�}1�@�����H���p�����[�^�ɑ��� ECs�y�ъ֘A����ύX�J�e�S���[�̓���̂��߂̃f�B�V�W�����c���[
�@�@�@�@�iICH Q12�K�C�h���C��������p�j
�@
�@Established Conditions
�iECs�j �Ƃ́C���F���ꂽ�i�ڂ̍H����\��i����ۏ���C�\���ɂ����Ē�`���ꂽ�C���i�̐������@�C�{�݂���ё��u�C�Ȃ�тɊǗ��헪�̗v�f�̋L�q�ł���CECs��ύX����ۂɂ�FDA�ɘA�����K�v���邪�C�V�����v���ł͂Ȃ��i�}2�j�B�Ȃ�ECs�ɂ͈ȉ��͈̔͂��܂ށB
�@�E�����܂̐����⎎���̎{��
�@�E�o�C�I���i�̏o�������̌�����K�i
�@�E�������@�i�H���Ǘ��C�H���̏��Ԃ��܂ށj�C���u�C�H���p�����[�^�Ɖ��͈�
�@�E����C���܁C���ԑ́C���̑��̕����̋K�i�y�ю������@
�@�E�e��y�ю{���n�C�ގ��C����т����K�i�y�ю������@
�@�E�P�����g���b�N�X���̈ێ����@

�}2�@Established Conditions
(ECs) FDA �K�C�_���X
�@
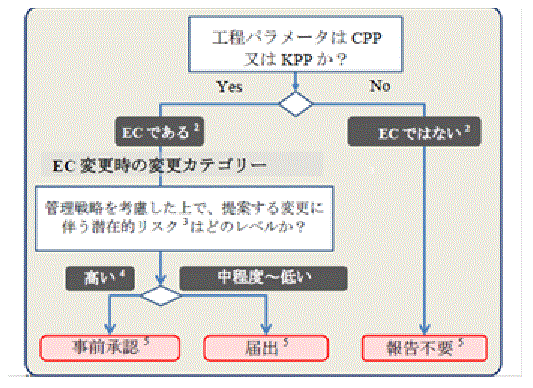
��EC�ƕύX�J�e�S���[�̌���@��3�j
�@EC�̌���ł́C���i�i���̊m�ۂ̂��߂ɊǗ�����K�v������p�����[�^���ۂ���EC�ɊY�����邩�f����B���Ȃ킿�C���̃p�����[�^��ύX�����ꍇ�̐��i�i���ւ̐��ݓI�ȃ��X�N�]���ɂ��C���O���F���K�v�ł��邩�͏o�����̂�����ł��邩�����肵�Ȃ���Ȃ�Ȃ��B�T�N���~������̃��b�N4�j���ނƂ��Č������C�{����ł͓��肳�ꂽ����CQA�̂����C�s�����̊Ǘ����Ƃ肠���Ă���B�T�N���~�����܂Ɍ��炸�C���̕s�����͐��܂̈��S���ɉe�����y�ڂ����߁C����CQA�̂Ȃ��ł����ɏd�v�Ȃ��̂ƈʒu�Â�����B
�@�����̃��X�N�]���ł́C�މ������ʁi����CQA�j�ɉe�����y�ڂ��\���̂���̈�̓�����s���C����Ȃ郊�X�N�]�������{�C����CQA�ɉe�����y�ڂ��\���̂���H���p�����[�^����肷��B�����āC�O�i�K�œ��肵���H���p�����[�^�̗މ������ʁi����CQA�j�֎��ۂɉe�����y�ڂ��̂�������B���̌��ʁC���������H���p�����[�^�����i�i���̊m�ۂ̂��߂ɊǗ�����K�v������p�����[�^���ۂ���EC�ɊY�����邩�f����B
�@����ɂ��̍��ڂɂ��āC�p�����[�^��ύX�����ꍇ�̐��i�i���ւ̐��ݓI�ȃ��X�N�]���ɂ��C���O���F���͂��o�����������肷��B�l�����ׂ������Ƃ��āC�{��������ł�Enhanced
Approach�i���i��@�j�ŊJ�������T�N���~������ɓ��Ă͂߂邱�ƂŁCEC�Ƃ��̕ύX�J�e�S���[���ǂ̂悤�Ɍ��肳��邩�������Ă���B
�@ EC�ł��莖�O���F���K�v�ȗ�
�@�T�N���~�����b�N�ł́CStep2�̌��������̗�p���x�Ɛ��̗ʂ�CPP�Ƃ��ē��肳�ꂽ�B���������āC�����2��ނ̍H���p�����[�^�́CEC�ɑ�������B�����̍H���p�����[�^�́C�����v��@�̌����̌��ʁC�f�U�C���X�y�[�X���m������Ă���B�G�^�m�[���̎��ʂɑ��鐅�̗ʂ̃f�U�C���X�y�[�X�́u20�`35���v�ł���B���F�\�����̋L�ڂ́u�G�^�m�[���̎��ʂɑ���20�`35���̎��ʂɑ������鐅����������C����0.15�`0.5���ŗ�p���C18���Ŋh�a����v�ƂȂ��Ă���B
�@�����̌��ʁC�����͌���CQA�ɉe�����y�ڂ��d�v�H���p�����[�^�iCPP�j�ł���C���������͈́i�f�U�C���X�y�[�X�j���s�K�����E�ɋ߂��i�����X�N�j���Ƃ����������B�܂�C�����̍H���p�����[�^�͂����͈̔͂���E����ƁC�����܂�����CQA�����Ȃ��Ȃ�Ƃ������Ƃł���B���̂��߁C�������̍H���p�����[�^�̊Ǘ��͈͂����̃f�U�C���X�y�[�X�͈̔͂Ɠ����ɂ��Ă��C�i���ɑ��郊�X�N�͑傫���܂܂ɂȂ�B���������āC�����̍H���p�����[�^�͎��O���F��L����EC�ƂȂ�Ɣ��f�����Ɖ�����ꂽ�B
�A EC�ł���͏o�ƂȂ��
�@Step1�ł̌��������̐��̗ʂ�CPP�ɓ��肳��Ă���CEC�ɑ�������B����@�ƈقȂ�̂͂��̍H���p�����[�^�̃f�U�C���X�y�[�X���C�u10�`50���v�Ɣ�r�I�L���_�ł���B
�@�\�����̋L�ڂ́u�G�^�m�[���̎��ʂɑ���25�`35���̎��ʂɑ������鐅�������ė�p���C20���Ŋh�a����v�ƂȂ��Ă���B�H���p�����[�^�͂��̃f�U�C���X�y�[�X����E����ƁC����CQA�͖������Ȃ��Ȃ�B�������C���̃P�[�X�̏ꍇ�C�f�U�C���X�y�[�X�͈̔͂���r�I�L�����߁C�������̊Ǘ��͈͂��f�U�C���X�y�[�X�ɑ��ď\�������͈́i25�`35���j�Őݒ肷�邱�Ƃ��ł���B���̂悤�ȊǗ��헪���Ƃ邱�Ƃɂ��C�i���ɑ�����ݓI���X�N��ጸ���邱�Ƃ��ł���B���̂��߁C�ύX�J�e�S���[�͎��O���F�ł͂Ȃ��͏o�ɊY������EC�ɂ��邱�Ƃ��ł���\��������B
�B EC�łȂ���i����CQA�ɉe�����y�ڂ��Ȃ��j
�@Step1�̌������ɂ������p���x�C�Y�����ԁC�h�a���x�C�ێ����ԁCTHF�Z�x�CStep2�̌��������ɂ�����C�Y�����ԁC�h�a���x�C���Y���܂ł̎��Ԃ͌���CQA�ɉe�����y�ڂ��Ȃ����Ƃ���CEC�ɂȂ�Ȃ��Ɣ��f�����Ƃ����B
�@��̒P�ʑ���̃A�E�g�v�b�g�͎��̑���̃C���v�b�g�ł��邽�߁CEC ����������ۂɂ́C�����H���ƊǗ��헪�S�̂̕�I�ȗ������K�v�ł���B
���ύX�J�e�S���[��
�@���@ ���O���F�iPrior Approval; PA�j-PAS, Type II, ���F�����ꕔ�ύX���F�\����
�@���@ �͏o�E�����X�N�̕ύX �iNotification Moderate; NM�j-CBE
30, Type IB, ���F�����y���ύX �͏o��
�@���@ �͏o�E��X�N�̕ύX �iNotification Low; NL�j-AR, Type
IA, ���F�����y���ύX�͏o��
�@���@�s�v �iNot Reported; NR�j
�R�DICH-Q12 ���i���C�t�T�C�N���}�l�W�����g�ƕύX�Ǘ��̐v����
1�jPACMP�iPost -Approval
Change Management Protocol:���F��ύX�Ǘ����{�v�揑�j�Ƃ́C�u���F�\�����ɗ\�肳��Ă��邠�邢�͑z�肳��Ă���s�̌�̕ύX�v���w���B
�@Step1
�@�]�����@��\�߃v���g�R�[����CTD��R�i�n��̗v���j�̍��Ɋ܂߂Ă����C���O�R����������B
�@Step2
�@���F��C�v���g�R�[���Ɋ�Â��ĕύX���s��ꂽ�ꍇ�ɂ́C�\�ߎ��O�Ɋ܂߂Ă������ύX�̃J�e�S���[�ɂ��C�ύX��implementation���\�ƂȂ�B
�@�Ⴆ�C���B�ł�Type�U����Type�TB�ł̕ύX���\�ɂȂ�C�č��ł�PAS����CBE-30
��CBE-0�C����ɂ͔N���ł̕ύX���\�ɂȂ�ꍇ������B
�i�Q�l�jProduct Life Cycle Management�iPLCM�j�h�L�������g�F�ύX�}�l�W�����g�Ɋւ��e��������������i�C�Ӂj�F
�E�Ǘ��헪�̊T�v�CECs�CEC��ύX����ۂ̕ύX�J�e�S���[�CPACMP��Post-approval
CMC�R�~�b�g�����g�Ȃǂ��܂߂�B
2�jPACMP��p�������F�����̕ύX���x
�@Q12�u���i�̃��C�t�T�C�N���}�l�W�����g�K�C�h���C���v�X�e�b�v3�i2018�N1���j�F
�@�������́C"Technical and Regulatory Considerations
for Pharmaceutical Product Lifecycle Management"�Ƃ����C�ύX�}�l�W�����g�̒��a��ړI�Ƃ��Ă���B
�@����́C���F��̕ύX�Ɋւ��ď_��ȉ^�p������Ă��Ȃ��v�������葱���C��o���������m�����C���F��̕ύX�̍s���葱���ɏœ_�Ă�B
�S�D���F��ύX�Ǘ��iPACMP�j�Ƃ��̐i�ߕ�
4.1�@PACMP �̗��p
�@PACMP �̗��p�ɂ́C��ʓI�Ɉȉ���2�̃X�e�b�v���K�v�ƂȂ�F
�@�X�e�b�v1�F��Ă���ύX�C���̍����C���X�N�}�l�W�����g�̊����C�ύX�̉e����]�����邽�߂ɒ�Ă��錟���y�є����C�������K�v�����邻�̑��̏����i���F���ꂽ�K�i�y�ю������@�ɕύX���Ȃ����Ƃ̊m�F���j�C���Y�ύX�ɑ��Ē�Ă���ύX�J�e�S���[�C���тɂ��̑��̗��t���ƂȂ���i�����Q�Ɓj���L�ڂ������{�v�揑�̐\���BPACMP
������CTD���W���[��3.2.R �Ɋ܂߂邱�Ƃ��ł���3�B���̎��{�v�揑�́C���s�ɐ旧���āC�K�����ǂɂ��R������C���F�����B
�@�X�e�b�v2�F���{�v�揑�Ɏ����������y�ь��������{�����B����ꂽ���ʁ^�f�[�^�����{�v�揑�ɂ��锻�������Ă���C���̑��̏������������Ă���ꍇ�CMAH
�͏��F���ꂽ���{�v�揑�ɋL�ڂ���Ă���J�e�S���[�ɏ]���Ă��̏����K�����ǂɒ�o���C�K�X�K�����ǂɂ��R������B�ύX�J�e�S���[�ɂ���āC�ύX�̎��{�O�ɋK�����ǂ̏��F���K�v�ȏꍇ�ƕs�v�ȏꍇ������B���{�v�揑�ɂ��锻���y�с^���͂��̑��̏����i�X�e�b�v1
�Q�Ɓj�����Ȃ��ꍇ�́C���̃A�v���[�`�𗘗p���ĕύX�����{���邱�Ƃ͂ł����C����Ɍ��s�̋K�����̓K�C�_���X�y�ъ֘A����ύX�J�e�S���[�ɏ]�����ƂɂȂ�B
�@�����̑����́u����PHARMSTAGE�v�Q�O�Q�T�N�S�����@�{���ł�������������
�@�@����PHARMSTAGE�̃z�[���y�[�W�͂�����
�@�@https://www.gijutu.co.jp/doc/magazine_pharm%20stage.htm
�Q�l����
1�j�uICH Q12 ���i�̃��C�t�T�C�N���}�l�W�����g
�K�C�h���C���i�āj�vSTEP 3�C����29�N
2�j�u���C�t�T�C�N���}�l�W�����g�ɂ�����ICH Q12�̎��H�Ɗ��p��vPHARM TECH
JAPAN Vol. 37�@No.6 �i2021�j83�i972�j
3�j��27����{PDA����w��N��i2020�N12��8���ɃI�����C���J�Áj
|


