|
�P�D�͂��߂�
�@2021 �N�ɉ���GMP �ȗ߂��{�s����C���{�ɂ����Ă����������h�~�[�u���������ꂽ�B����ɂ��C�w�I����ѓŐ��w�I�]���Ɋ�Â��Ȋw�I�f�[�^�����Ƃɐݒ肳�ꂽ�c�����x�l���g�p�������o���f�[�V�����̎��{�����߂���悤�ɂȂ����B���̍ہC�Q�l�Ƃ����PIC/S�i���j�֘A�K�C�_���X�����ɂ�HBEL�iHealthbased
exposure limit, ���N�ւ̉e���Ɋ�Â����I���E�j�K�C�h���C��1�j���܂܂�Ă���C���̕����ł͓Ő��w�I�]���Ɋ�Â�PDE�iPermitted
Daily Exposure�C������I���e�ʁj�̐ݒ���@���K�肳��Ă���B
�@���������āC���{�����ɂ����Ă��CPDE ���\�Ƃ���HBEL �Ɋ�Â����o���f�[�V�����̎��{���CGMP�ɂ�������������h�~�v���������߂ɕK�{�ł���B
�@�{�e�ł́CPDE �̎Z�o���@�����PDE �ݒ背�|�[�g�̍쐬�ɂ��ĉ������B
�@�� PIC/S : Pharmaceutical Inspection ConventionPharmaceutical
Inspection Co-Operation Scheme�i���i���@����y�ш��i���@�����X�L�[���j
�Q�DPDE �Z�o
�@PDE ���Z�o�����Ƃ́C�������̃X�e�b�v��Ői�߂���i�}1
�Q�Ɓj2�j�B�܂��C�@ �L�Q���̓���ɂ����āC����\�Ȃ��ׂĂ̔�Տ�����їՏ������f�[�^����L�Q������肷��B�����ł̒��ӓ_�́C��p���l�ɂƂ��ėL�Q�ȕω��Ɋ܂܂��Ƃ����_�ł���B���ɁC���̗L�Q���̒�����C�A
�d��ȉe���̓�����s���B�Տ������f�[�^��p����ꍇ�́C�ł���p�ʂŔF�߂���L�Q��p��I�����C��Տ������f�[�^�̏ꍇ�͖�p��q�g�ւ̊O�}�����l������B
�@����ɁC�B POD�iPoint of Departure�C�Ő��w�I�o���_�j�̑I�����s���B�����̏ꍇ�C�e�����F�߂��Ȃ��ō��p�ʂł���NOAEL�iNo
Observed Adverse Effect Level�j�܂���NOEL�iNo Observed
Effect Level�j��I�����邪�C����炪�����Ȃ��ꍇ�ɂ́C�e�����F�߂���ŏ��p�ʂł���LOAEL�iLowest
Observed Adverse EffectLevel�j���邢��LOEL�iLowest Observed
Effect Level�j��I������B�܂��C���������ƃq�g�Տ������œ��l�̉e�����F�߂��Ă���ꍇ�ɂ́C�Տ������f�[�^���d������B�q�g�ŕ]���ł��Ȃ�������B�����Ő��Ȃǂɂ��ẮC��Տ������f�[�^����POD
��I������B
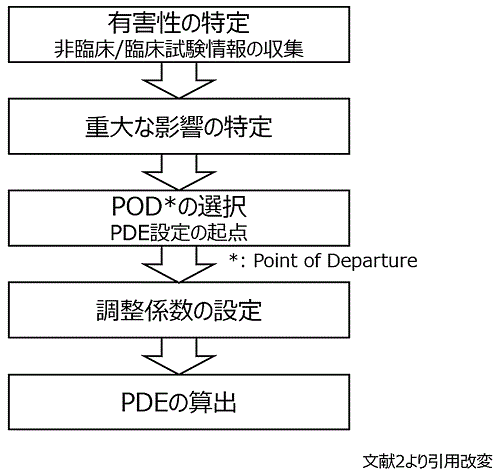
�@�@�}1�FPDE�ݒ�̗���
���ɁCPOD �̎����f�[�^�Ɋ�Â��āC�C
�����W���̐ݒ�iF1 �` F5�C�o�C�I�A�x�C���r���e�B��W�����j���s���B�����̒����W���́C���̖�܂����POD
�Ɋւ��邳�܂��܂ȕs�m�����ɑΉ����邽�߂ɐݒ肳���B�Ō�ɁC�v�Z���Ɋ�Â��C�D PDE �̎Z�o���s���BPDE
�̌v�Z���͈ȉ��̂Ƃ���ł���B
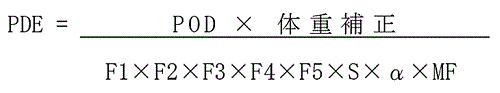
�@�@�@POD�F Point of Departure, PDE �Z�o�̂��߂̓Ő��w�I�ȏo���_
�@�@�@F1 �` F5�F �l�X�ȕs�m�������l�����邽�߂̒����W��
�@�@�@F1�F��ԍ�
�@�@�@F2�F�̍�
�@�@�@F3�F�������Ԃ̒���
�@�@�@F4�F�e���̏d�含
�@�@�@F5�FLOAEL ����NOAEL �ւ̊O�}
�@�@�@S �F����Ԃ̌W���܂��͒~�ϐ��̌W��
�@�@�@�� �F ���I�o�H�O�}�̂��߂̃o�C�I�A�x�C���r���e�B��W��
�@�@�@MF �F ���̑��̒����W���i�f�[�^�x�[�X�̊��S�����j
2.1�@�L�Q���̓���
�@����\�Ȃ��ׂĂ̔�Տ������C�Տ��������̓Ő�������肵�������ŁC�L�Q���̓�����s���B���̖�܂��q�g�ɑ��Ď��Ő��̑S�̑���c�����邱�Ƃ͏d�v�ł���C��{�I�ɂ̓q�g�ɊO�}�\�ȓŐ����d������B����������ɓ��ٓI�ȕa�ԓ��̓Ő����ɂ��ẮCPDE�ݒ�̍ۂɗ��p���Ȃ��B
2.2�@�d��ȉe���̓���
�@���肳�ꂽ�L�Q���̒��ŁC�q�g�ɊO�}�\�ł���C���ł���p�ʂœŐ�������������̂��d��ȉe���Ƃ��ē��肷��B���̍ہC�e�����t�I���ǂ����̊ϓ_���d�v�ł���C�s�t�I�ȓŐ��͂��d��Ƃ݂Ȃ����B���ɏd�v�ȓŐ��ɂ́C�����C���B�����Ő��C�_�o�Ő������܂܂��B
2.3�@POD �̑I��
�@�d��ȉe���̒�����CNOAEL�i�e�����F�߂��Ȃ��ō��p�ʁj��LOAEL�i�e�����F�߂���ŏ��p�ʁj�C���邢�̓x���`�}�[�N�h�[�X�@�ɂ��BMDL10�i���邢��BMDL05�j�̒l��POD�iPoint
of Departure�C�Ő��w�I�o���_�j�Ƃ��đI�����CPDE ���Z�o����B���̍ہC��蒷���Ԃ̎����̃f�[�^��D�悷��B���l�̃G���h�|�C���g���Տ������Ɣ�Տ������ŔF�߂�ꂽ�ꍇ�͗Տ��������ʂ�D�悷�邪�C�Տ������̎������Ԃ��������Z���ꍇ�́C��Տ������f�[�^��POD
�̌��Ƃ��Č�������B
�x���`�}�[�N�h�[�X�@�ɂ���
�@�x���`�}�[�N�h�[�X�@�ł́C�����f�[�^�ɗp�ʔ����Ȑ��Ă͂߂�POD ���Z�o����B�x���`�}�[�N�p�ʁiBMD�j�́C����̌��N�e���̔����������̐����w�I�����̕ω��i�x���`�}�[�N�����CBMR�j�Ɋ֘A����p�ʂł���B����BMR
�ɑ���95���M�����E�̉����l��BMDL �Ƃ��ĎZ�o�����B
�@���p�\�ȃf�[�^�̎����\���ɍ����ꍇ�ɂ́C�x���`�}�[�N�h�[�X�@��p����BMDL10�i���邢��BMDL05�j���v�Z���C������Ȋw�I�ɑÓ���NOAEL
�Ƃ��ėp���邱�Ƃ��ł���3-5�j�BNOAEL ��LOAEL �́C���������Ŏ��O�ɐݒ肳�ꂽ�p�ʂɈˑ�����l�ł��邽�߁CBMDL
�̕����d�v�ȉe���̗p�ʔ����������K�ɑ�\���Ă���\��������B BMDL ��POD �Ƃ��ėp����ꍇ��F5
�̒����W���͕s�v�ł���B
2.4�@�̏d�̕
�@��Տ�������POD ��I�������ꍇ�́CPOD
���̑̏d������̗p�ʁimg/kg/day ���j�ɕ���C�q�g�̑̏d��50kg �Ƃ��čČv�Z����1�j�BPOD
���q�g�ւ�1 �����^�ʂœ����Ă���ꍇ�́C�̏d��͕s�v�ł���B
2.5�@�����W���̐ݒ�
�@�l�X�ȕs�m�����ɑΉ����邽�߂̒����W����ݒ肷��B���̍ہC���ꂼ��̒����W�������S�ɓƗ����Ă���킯�ł͂Ȃ��ꍇ�����邽�߁C�d�����Ē����W����K�p���Ȃ��悤�ɒ��ӂ��K�v�ł���B��ʂɁCF1
�` F5 �̒����W���̐ς�5,000 �` 10,000 �ȏ�ɂȂ�ꍇ�́C�s�m���������ɍ����CPDE
�̎Z�o�͓K�ł͂Ȃ��Ƃ���Ă���B
�i1�jF1�F��ԍ�
�@PDE �́C�q�g�̃f�[�^�Ɋ�Â��Đݒ肷�邱�Ƃ��]�܂������C�d��ȉe���ɂ��ăq�g�̃f�[�^�������Ȃ��ꍇ��C�Տ������̎��{�O�ɂ́C��Տ������f�[�^��POD
�Ƃ��đI�肷��6�j�B���̍ہC��������q�g�ւ̊O�}���s�����߁C�퍷�Ɋ�Â���ӂ���w�I�ȃp�����[�^�[�̈Ⴂ���l�����C�K�Ȓ����W����ݒ肷��K�v������B
�@�`���I�ɂ́C���̎퍷���l�����������W���Ƃ��āC�S�Ă̓�����ɑ��ăf�t�H���g�l10 ���K�p�����B����10
�Ƃ����f�t�H���g�l�́C�g�L�V�R�L�l�e�B�N�X�iTK�j��4.0�C�g�L�V�R�_�C�i�~�N�X�iTD�j��2.5
�ɕ��������7,8�j�B
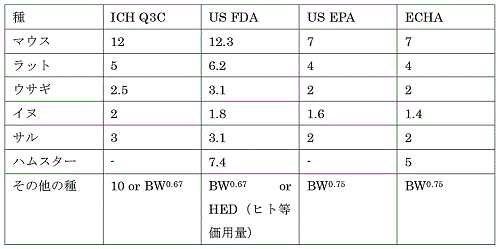
�@�ߔN�̃K�C�h���C���i�\1�j�ł́C�퍷�̒����͎��̎��Ɋ�Â��A�����g���b�N�E�X�P�[�����O�ōs����B
�@�\�P�@�e�@�ւɂ��K�C�h���C���ɂ����Đ�������Ă����ԍ��̒����W��9-12�j
�@
�܂��C�퍷�������������ŗL�̃g�L�V�R�L�l�e�B�N�X�iTK�j�y�уg�L�V�R�_�C�i�~�N�X�iTD�j�f�[�^�����݂���ꍇ�́C�����ŗL�̒����W���iCSAF,
ChemicalSpecific Adjustment Factor�j �̎Z�o����������Ă���8,13�j�B
�i2�jF2�F�̍�
�@�̊ԕϓ��ɑΉ����钲���W���́C�q�g�W�c���̃g�L�V�R�L�l�e�B�N�X�iTK�j����уg�L�V�R�_�C�i�~�N�X�iTD�j�̕ϓ����l�����C�u�d��ȉe���v�����肷�邽�߂Ɏg�p���ꂽ�����W�c�Ɣ�r���āC�N��
�E ���� �E ������ �E ��`���̏����ɂ��e�����₷���q���ȃT�u�O���[�v��ی삷�邽�߂ɐݒ肳���W���ł���B
�@����F2 �W���́C�ʏ�f�t�H���g�l�Ƃ���10 ���p�����CTK ��TD �̒����W���́C���ꂼ��f�t�H���g�l�Ƃ���3.16
�ɕ��������7�j�B������TK, TD �̒l�́C���w�����ŗL�̒����W���iCSAF�j�ɒu�������邱�Ƃ��\�ł���B
�@CSAF �̎Z�o�ɂ����āCTK �ɂ��Ă͌����Z�x�̎��ԋȐ����ʐρiAUC�j�܂��͍ō��������Z�x�iCmax�j��p���邱�Ƃ��ł���B���L��������Ő�������C�s�t�I�ȑ����������N�����Ő������ɂ��ẮCAUC
�̂悤�Ȍo���I�ȗp�ʂ̐ϕ��w�W���ł��K�Ƃ���CPDE��p����ꍇ�̒�p�ʈ�̃��X�N�]���ɂ����Ă�AUC���D�悳��邱�Ƃ������B����C�Ő������ɂ�����Cmax
���d�v�ƂȂ�e�����݂��镨���i���Ƃ��C�A�~�m�O���R�V�h�n�R��������C�S�Ő���_�o�Ő��������j�ł́CCmax
���D�悳���ꍇ������BPD �ɂ��ẮC�ʏ�͗��p�\�ȏ���Ȃ����߁C�f�t�H���g�l�ł���3.16
����ʓI�Ɏg�p�����6�j�B
�@����ҏW�c����ё�\�I�Ȋ��ҏW�c�̃f�[�^����̓��o�����CSAF �́C�ȉ��̎��ŕ\�����6�j�B
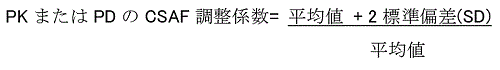
�܂��C���̍����T�u�O���[�v�����肳��C���̃f�[�^������\�ł���C����ȕ����W�c�Ɗ��̍��������W�c�̓�����̕��z�Ƃ݂Ȃ��C�ȉ��̎��ŕ\�����Ƃ��ł���6,14,15�j�B
�@

�i3�jF3�F�������Ԃ̒���
�@PDE �͈ꐶ�U�������I���Ă����N�ɉe���̂Ȃ��p�ʂƂ��Đݒ肳��邽�߁CPOD �Ɏg�p���ꂽ�����̔��I���Ԃɑ���s�m����������K�v������B�ʏ�C��蒷���̔��I�Ŋώ@�����NOAEL
�͒Z���̎��������������Ȃ�X��������B�܂��C�Z���Ԃ̔��I�ł͔��������C�����Ԃ̔��I�ł̂݊ώ@�����e��������\��������B
�@���̎������Ԃ̒����W���iF3�j�́C�����̎�ށE���I�V�i���I�C��Տ������ɂ����Ďg�p���ꂽ������̎����ȂǂɈˑ�����B�\2
�́C�e�@�ւ̃K�C�h���C���Ő�������Ă���F3 �̒����W���������Ă���B
�\�Q�@�e�@�ւɂ��K�C�h���C���ɂ����Đ�������Ă��鎎�����Ԃ̒����W��12,16,17�j

�@�����̋@�ւł́C���}�������i�ʏ�28 ���Ԏ����j���疝�����I�ւ̒����W����6 �` 10�C�����������i�ʏ�90
���Ԏ����j���疝�����I�ւ̒����W����2 �` 3 �Ƃ���Ă���BICH Q3C/3D �ɂ����ẮC���B�����Ő������Ŋ튯�`�����S�̂��J�o�[���鎎�����Ԃł���CF3�����W����1
�Ƃ���Ă���B�q�g�̎���Ƃ��āCICHQ3D �ł́C�o�i�W�E���ɑ���3 ���������̔��I���Ԃ�5�C�R�o���g�ɑ���3
�����̔��I���Ԃɂ�2�C��ɑ���2 �N�ȏ�̔��I���Ԃɂ�1 �̒����W�����ݒ肳��Ă���18�j�B
�\3�@PDE�ݒ茟�����Ă���F3�����W���i��Տ������y�уq�g�̎���j2�j
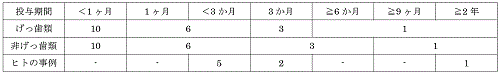
�@
�@PDE �ݒ茟����ł́C�����̏�������F3
�W����������2�j�B
�@Naumann ��Weideman �́C����ꂽ���̍����f�[�^�Z�b�g�Ɋ�Â��āC��������NOAEL
�Ɩ�����NOAEL���r���������̕��͂���C ������NOAEL ��NOAEL �֕ϊ�����ۂ̒����W���Ƃ��čœK�Ȑ���l��3
�ƒ�Ă��Ă���4�j�B�܂��C��܂�TK �y��TD ���l�����Ď������Ԃ̒������s���ꍇ������B
�i4�jS�F����Ԃ̌W���C�܂��͒~�ϐ��̌W��
�@PDE ��P�^�܂��͒Z���Ԃ̎����Ɋ�Â��Đݒ肷��ꍇ�C�����ŗL�̐����~�ϐ���TK �f�[�^�Ɋ�Â��Ē���ԌW���gS�h��p���邱�Ƃ��ł���B����Ԃ̑̓��Z�x�́C1
�R���p�[�g�����g���f���ł͏�����������p���āC�܂��̓}���`�R���p�[�g�����g���f���ł͏����Ȑ��̏I�����̔������C���邢�͂��̑��̖��ԃf�[�^��p���Čv�Z���邱�Ƃ��ł���B�̏����������iT1/2�j���P�^�܂��͒Z���Ԃ̎����ł킩���Ă��邪�C����Ԃ̃v���t�B�[�����s���ȏꍇ�C����Ԃ��B�����ꂽ�Ƃ��̌������Z�x�ƁC�P�^�̌������Z�x�̔�Ƃ��āC������p���Ė̒~�ς̒��x�𐄒肷�邱�Ƃ��ł���13�j�B
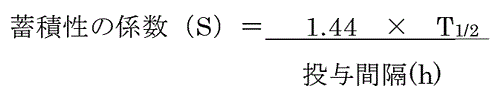
�@�~�ϐ��̌W����PDE �ɓK�p����ہC���^�Ԋu��POD�̓��^�Ԋu�ł͂Ȃ��CPDE
�̌��������V�i���I�ɂ����锘�I�Ԋu�i�ʏ�24 ���ԁj��p����B��������������r�I�����i24
���Ԉȏ�j�ł́C�����J��Ԃ����I������ɒ���Ԃ̌������Z�x�ɓ��B����B���̂��߁C�Z���Ԃ̎������瓾��ꂽPOD
��p����ꍇ�C����Ԃ̉e�����s���Ȃ��߁CS �ɂ���ē��^�ʂ�������K�v������B ����C �ێ����^�ʂł̒���Ԃ����炩�ł���CPOD
�̎������Ԃ��\�������ꍇ�́CS �̒����͕s�v�ƂȂ�19�j�B
�@���̒~�ϐ��̌W���iS�j��F3 �̒����W���͊֘A���Ă���C�Ɨ������Ă��Ȃ����߁CS ���g�p����ۂɂ́CF3�Ƃ̏d���ɒ��ӂ��K�v�ł���B�ʏ�CF3
��p���邱�Ƃ�S ���l������邱�Ƃ��������CS ��K�p����ۂɂ�F3 �������Ďg�p���邱�Ƃ��K�Ƃ����13�j�B���������������ɒZ����܂̏ꍇ�́C�~�ϐ��̌W����1
�ȉ��ɂȂ邽�߁CF3 �ɂ�����TK �̍l�����s�v�ɂȂ�\��������B�������CTD �͕ʓr�l������K�v�����邽�߁C��͊w�I�~�ς̉\�����܂މe���̍�p�@�����l������F3
��ݒ肷��K�v������B
�i5�jF4�F�e���̏d�含
�@POD �ɐݒ肵���e�����C�d�ĂȓŐ��ł���ꍇ�ɂ�F4 �����W�����K�p�����B�Ⴆ�C��`�Ő���Ȃ������C�_�o�Ő��C�Ê�`����C���邢�͕s�t�I�ŏd�Ăȉe���i��F�d�ĂȊ̏�Q�C�Ԏ����x���C���S�j������ꍇ�ɓK�p����邱�Ƃ�����B
�\�S�@�e�@�ւ̃K�C�h���C���ɂ�����F4�����W��2,9,18�j

�@
�@ICH Q3C/3D �ł́C��`�Ő���Ȃ������Ɋւ��āC���̏؋��̒��x�ɉ����āC5
�` 10 ��F4 �����W����ݒ肵�Ă���9,18�j�B
�i6�jF5�FLOAEL ����NOAEL �ւ̊O�}
�@POD �Ƃ��Ďg�p���������̌��ʂ�LOAEL �܂���LOEL �ł������ꍇ�CNOAEL�i�܂���NOEL�j�ւ̊O�}���K�v�ƂȂ�B���̏ꍇ�C�K�Ȓ����W����K�p����NOAEL
�𐄒肷�邱�Ƃ����߂���B���̒����W���͉e���̎�ނ�d�Ă��C�p�ʔ����Ȑ��̌X���ȂǂɊ�Â��ăP�[�X�o�C�P�[�X�Őݒ肷��K�v������B
�@���Ƃ��CLOAEL �̉e�����t�I�Ȗ�p�Ɋ�Â����̂ł���CF5 ���������ݒ�ł���\��������B����C����̉ȂǏd�Ăȉe�����F�߂���ꍇ�ɂ́C���傫�ȌW�����K�v�ƂȂ�20�j�B�܂��CPOD
����p�Ɋ�Â����ł��Ⴂ�Տ���p�p�ʂł���ꍇ�ƁC�R����܂Ȃǂ̍ő�ϗʂ����Ƃɐݒ肳���p�ʂ̏ꍇ�ł́CF5
�ɐݒ肷�ׂ��W���͑傫���قȂ�B
�@�e�@�ւ̃K�C�h���C�����ł́C3 �` 10 �̌W������������Ă���B�e�@�ցE���҂�̐�������l��\�Ɏ����B
�\�T�@�e�@�ւ̃K�C�h���C���y�ђ��҂ɂ�����F5�����W��4,9,11,12,19)
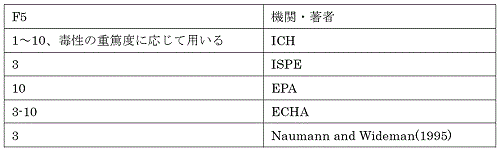
�@
Naumann �� Weideman �́C�������̎��̍����f�[�^�Z�b�g�̉�͂Ɋ�Â��CLOAEL
����NOAEL �ւ̒����Ɏg�p����W���Ƃ��čŗǂ̐���l���C3�i100.5�j�ƒ�Ă��Ă���4�j�B
�i7�j ���F���I�o�H�O�}�̂��߂̃o�C�I�A�x�C���r���e�B�̕�W��
�@PDE ��ݒ肷��ۂɁC���I�o�H���قȂ�POD ���g�p������Ȃ��ꍇ�C�o�H�Ԃ̊O�}���K�v�ƂȂ�BPDE
��ݒ肷��o�H�̃o�C�I�A�x�C���r���e�B�iBAPDE�j�ƁCPOD �̃o�C�I�A�x�C���r���e�B�iBAPOD�j�������Ă���ꍇ�C�o�H�ԊO�}�̌W���͈ȉ��̎��ł���킳���2,4,13�j�B
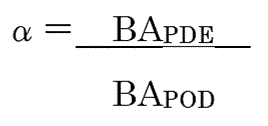
�@ICH Q3D �ł́C���˂�z���o�H�̃f�[�^���Ȃ��ꍇ�C�o�����I���̃o�C�I�A�x�C���r���e�B�iBA�j����ɏC���W����K�p���C�o�����^��PDE
�l���璍�˂���ыz���o�H��PDE �l���Z�o���Ă���18�j�B
�\�U�@ICH Q3D�ɂ�����BA�̏C���W��18�j

�Q�l����
1�jPharmaceutical Inspection
Convention Pharmaceutical Inspection Co-Operation
Scheme (PIC/S). Guideline on Setting Health Based
Exposure Limits for Use in Risk Identification
in the Manufacture of Different Medical Products
in Shared Facilities. (2018)
2�jPDE�ݒ茟����C�[������w��HBEL�I���p�{�݂ɂ�������������h�~�̂��߂�PDE�ݒ�C������Ђ��ق��i2021�j
3�jASTM, Standard Guide for Derivation of Health-Based
Exposure Limits (HBELs), E3219-20, (2020)
4�jB. D. Naumann and P. A. Weideman, Scientific
basis for uncertainty factors used to establish
occupational exposure limits for pharmaceutical
active ingredients. Human and Ecological Risk
Assessment. 1 (5) : 590-613 (1995)
5�j�L�����F�C���Y���C�x���`�}�[�N�h�[�X��@�̓K�p�̌���Ɖۑ�|���������f�[�^�ւ̓K�p�𒆐S�Ɂ|�C�Y�ƈ�w���r���[�CVol.
34�CNo. 1�C�i2021�j
6�jSargent E. V. et al., Guidance on the establishment
of acceptable daily exposure limits (ADE) to support
Risk-Based Manufacture of Pharmaceutical Products.
Regul. Toxicol. and Pharmacol. 65 : 242-250 (2013)
7�jRenwick, A. G., Data-derived safety factors
for the evaluation of food additives and environmental
contaminants. Food Addit. Contam 10, 275e305.
(1993)
8�jIPCS, Chemical-specific adjustment factors for
interspecies differences and human variability
: Guidance document for use of data in dose/concentration
response assessment. In: International Programme
on Chemical Safety (IPCS). World Health Organization
(WHO), Geneva. (2005)
9�jICH, Q3C(R9) Impurities: Guideline for Residual
Solvents, In: International Conference on Harmonisation
(ICH), Geneva. (2024)
10�jUS FDA (Food and Drug Administration), Guidance
for Industry,Estimating the Maximum Safe Starting
Dose in Initial Clinical Trials for Therapeutics
in Adult Healthy Volunteers. US Department of
Health and Human Services, Food and Drug Administration,
Center for Drug Evaluation and Research. (2005)
11�jUS EPA, A Review of the Reference Dose and
Reference Concentration Processes. Final Report
: EPA/630/P-02/002F. U. S. Environmental Protection
Agency (EPA), Washington, DC. (2002)
12�jECHA, Chapter R. 8 : Characterisation of dose
[concentration]- response for human health. In
: Guidance on Information Requirements and Chemical
Safety Assessment. Version 2.1. European Chemicals
Agency (ECHA), Helsinki, Finland, (2012)
13�jReichard, J. F., Maier, M. A., Naumann, B.
D., Pecquet, A. M., Pfister, T. E. P., Sandhu,
R., Sargent, E. V., Streeter, A. J.,Weideman,
P., Toxicokinetic and toxicodynamic considerations
when deriving health-based exposure limits for
pharmaceuticals�B Regul. Toxicol. Pharmacol. Volume
79, S67-S78, (2016)
14�jSilverman, K. C., Naumann, B. D., Holder, D.
J., Dixit, R., Faria, E. C., Sargent, E. V., Gallo,
M. A., Establishing data-derived adjustment factors
from published pharmaceutical clinical trial data.
Hum. Ecol. Risk Assess. 5, 1059-1089. (1999)
15�jNaumann, B. D., Silverman, K. C., Dixit, R.,
Faria, E. C., Sargent, E. V., Case studies of
categorical data-derived adjustment factors. Hum.
Ecol. Risk Assess. 7, 61-105. (2001)
16�jICH, S4 : duration of chronic toxicity testing
in animals (rodent and non rodent toxicity testing),
In : International Conference on Harmonisation
(ICH), Geneva. (1998)
17�jICH, M3 (R2) : guidance on nonclinical safety
studies for the conduct of human clinical trials
and marketing authorization for pharmaceuticals,
In : International Conference on Harmonisation
(ICH), Geneva. (2009)
18�jICH, Q3D(R2) : Guideline For Elemental Impurities,
In : International Conference on Harmonisation
(ICH), Geneva. (2023)
|


