|
�P�D�͂��߂�
�@�V��́C�ʏ퐻���̔����F��܂�10-15�N�Ƃ��������������K�v�ƂȂ�C���F�邽�߂ɕK�v�ƂȂ�i�R���j�����́C����10�N�ȏ���O�Ɏ擾���ꂽ�f�[�^�Ƃ������ƂɂȂ�B�܂�C�擾��10�N�ȏ�̌����ď��߂āC���̎��������{���������⌤�����Č��ł�����̂ƂȂ��Ă��邩�ǂ���������C����Ɏ������Ή����Ă��Ȃ��Ə��F�擾�ɏd��ȉe�����o�邱�ƂɂȂ�B�������C���낢��ȕω����\�z�����10�N��15�N��̂��Ƃ܂őz�肵�đΉ����Ƃ邱�Ƃ́C��X�l�ԂɂȂ��Ȃ��ł��邱�Ƃł͂Ȃ��B�ɂ�������炸���i�J���ɂ͂��ꂪ���߂��Ă���̂ł���C���ꂪ�{�e�̃e�[�}�ł���g�f�[�^�̕i���Ƃ��̐��f�[�^�̎����E�ۊǁh�Ƃ������ƂɂȂ�B���ɁCCMC
�iChemistry�C Manufacturing and Control�j�Ɋւ��J�����̃f�[�^�E�����ɋ��߂��Ă���̂��g�M�����̊�h1�j�ł���B���́g�M�����̊�h�́C�ߋ��ɋN��������Q�����������Ƃ��Ē�߂�ꂽ���̂ł��邪�C���̊�ɏ������Ă��Ȃ������́C���F�\�����̐R�������Ƃ��Ă͍̗p����Ȃ��B���̂��߁C���̊�̓��e���\���ɗ���������ŁC�e��f�[�^���擾�E�쐬�E�ۊǂ��邱�Ƃ��C���i�J���ɂ͕K�{�̗v���ƂȂ��Ă���B�{�e�ł͒��҂�̌o�������CMC�f�[�^�̎擾�E�ۊǂ̃|�C���g�ɂ��ďЉ��B���Ȃ݂Ɂg�M�����h�Ƃ́C�g�A�C�e�����^����ꂽ�����ŋK��̊��Ԓ��C�v�����ꂽ�@�\���ʂ������Ƃ��ł��鐫���h�iJIS�j�C���̒�ʓI�Ȏړx�ł���M���x
�iReliability�j�́C�g�A�C�e�����^����ꂽ���ԗ^����ꂽ�������ŋ@�\������m���h�iJIS�j�ƒ�`�����2�j�B
�@�Ȃ��C�\�������́C�ȉ��̎��_����R������邱�Ƃ��C��R�̎���3�j�ŏЉ��Ă���B
�@�E�@ ���{���ꂽ�������o���ꂽ�����̐M�������S�ۂ���Ă��邩�H
�@�E�@ �ړI�Ƃ�����\�E���ʂ��l�����C�K�Ƀf�U�C�����ꂽ�Տ����������{����C�ΏۏW�c�ɂ�������i�Ƃ��Ă̗L������������Ă��邩�H
�@�E�@ ����ꂽ���ʂɗՏ��I�Ӌ`������Ɣ��f�ł��邩�H
�@�E�@ �C�O�������܂߂Ď����S�̂łǂ̂悤�ȗL�Q���ۂ��o�Ă��邩�H
�@�E�@ ���X�N�Ɍ��������x�l�t�B�b�g�����҂ł��邩�H
�@�E�@���X�N������ł�������邩�H
�Q�D���j����U��Ԃ鏳�F�\���ɌW��f�[�^�̐M����
�@����܂łɐ\���f�[�^��GMP���ł̐����ɂ����āC�f�[�^�̐M�����ɌW�邢�����̖�莖�Ⴊ�������Ă���B�\1�ɑ�\�I�Ȏ���4�j�����������C�����ł͂�����������̂��������Љ��B
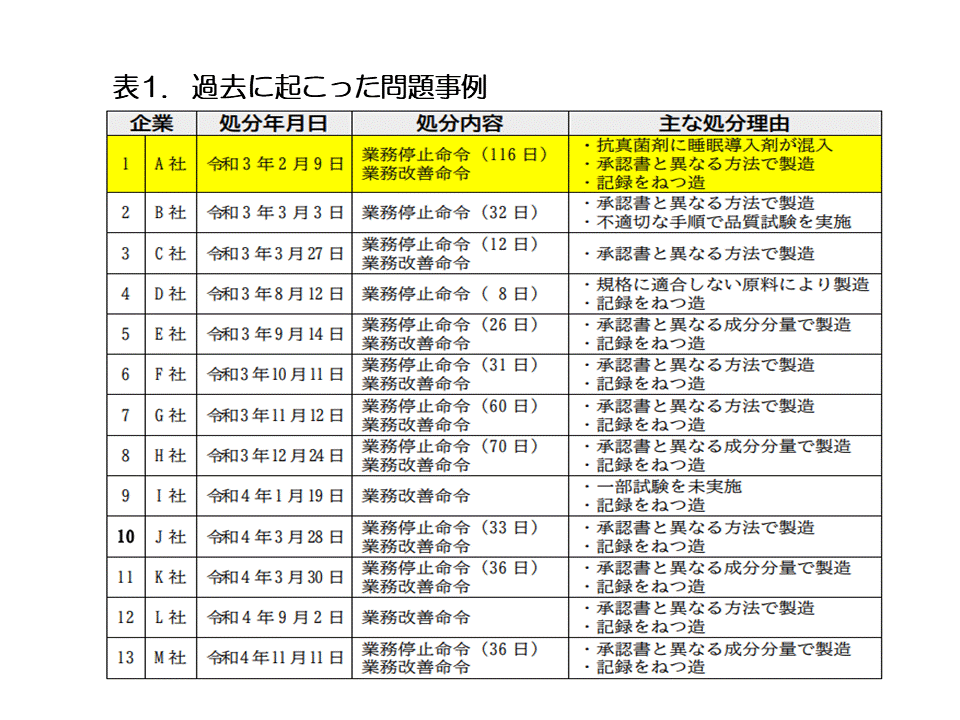
�R�D���i�J���ɂ�����M������ƐM�����̊
�@���i�J���ɂ����āC�����f�[�^�̐M�����͋ɂ߂ďd�v�ł���C���łɏЉ���悤�ɁC�f�[�^�̐M�����m�ۂ̋K�����݂����Ă���B���{�ɂ����ẮCCMC������Ȃǂ�ΏۂƂ��������ɑ���1997�N�Ɂg�M�����̊�h�����肳��Ă���B���́g�M�����̊�h�͓��{�Ǝ��̋K���ł���C�č����ł͕ʂ̌`�i�Ⴆ�CData
Integrity�j�Ńf�[�^�̐M�������S�ۂ���Ă���B���̂��߁C�č��Ƃ̋��c�̏�Łg�M�����̊�h�ƌ����Ă���������Ȃ��B���̏ꍇ�C���҂�́gGLP�̎菇��Data
Integrity�����킹�����́h�Ɛ�������悤�ɂ��Ă���B����GLP�i���i�̈��S���Ɋւ����Տ������̎��{�Ɋւ����j�C������GCP�i���i�̗Տ������̎��{�Ɋւ����j�́CICH�̃K�C�h���C���Ƃ��Ă��ʒm����Ă���C�O���[�o�����[���ł���B���́g�M�����̊�h�CGLP�CGCP�����킹�āC�g�M������h�ƌĂԂ��Ƃ������B�Ȃ��C�g�M������h�ɂ́C���̑���GPSP�i���i�̐����̔���̒����y�ю����̎��{�̊�j�Ȃǂ��܂܂��B
�@�����̋K���̖ړI�₻�̓��e�𗝉����邱�Ƃ́C���i�J�����v��ʂ�ɐi�߂邽�߂ɂ͕K�{�̗v���ł���C�����ł́CCMC�ɓK�p�����g�M�����̊�h�𒆐S�ɉ������B
�E�M�����̊
�@�g�M�����̊�h�́C1997�N�ɓ��{�Ő��肳�ꂽ���{�Ǝ��̋K���ł���B���̊�́C�\���u�W�������Ȃǂ̖�Q�������_�@�ɐ��肳��CGLP�̏ȗ߉��Ɠ������Ɏ{�s���ꂽ�B��Տ������ɂ����āC���S�������ɂ�GLP���K�p��������ŁCGLP���K�p����Ȃ�������CMC�֘A�����ɂ́g�M�����̊�h���K�p����邱�Ƃ���߂�ꂽ�B���̊�́CGLP���K�p����Ȃ������̈�ŁC�����f�[�^�̐M������S�ۂ��邽�߂ɐ݂����Ă���B�����v�揑�̍쐬�⎎�����ʂ̋L�^�E�ۑ��C�Č����̊m�ۂȂǂ����̓��e�Ɋ܂܂�Ă���C��Տ������ɂ�����f�[�^�̐��m���ƐM������ۏ��邽�߂̎w�j����Ă���B��@�@�{�s�K����43���́C�g�M�����̊�h�̗v�����ȉ��̂悤�ɒ�߂Ă���B
1�D�����̌v��E���{�E��
�@ �M�����̊�́C�����̌v�旧�Ă�����{�C�����Č��ʂ̕Ɏ���S�ߒ��ɂ����āC�f�[�^�����m�ōČ���������C�M���ł�����̂ł��邱�Ƃ����߂Ă���B����ɂ��C�����̊e�i�K�Ő��������f�[�^���K�ɊǗ�����C�������ʂ����m�ɔ��f����邱�Ƃ��ۏ����B
2�D�f�[�^�̋L�^�ƕۑ�
�@ �����f�[�^���K�ɋL�^����C�ۑ�����邱�Ƃ��K�肳��Ă���B�M�����̊�ł́C�f�[�^���������������C������邱�ƂȂ��L�^����邱�Ƃ����߂���B����ɂ��C�\�����ɒ�o���ꂽ�f�[�^�̐M�������m�ۂ���C���̑Ó������ؖ������B
3�D�Č����̊m��
�@ �M�����̊�́C�Č����̊m�ۂ��d�v�����Ă���B�������ēx���{���ꂽ�ꍇ�ł����l�̌��ʂ�������悤�ɐv����C���{����Ȃ���Ȃ�Ȃ��B
4�D�f�[�^�̓�����
�@�@�������@�⌋�ʂ����m�Ɏ�����C�K�v�ɉ����đ�O�҂����̃f�[�^�����ł���悤�ȏ�ԂłȂ���Ȃ�Ȃ��B����ɂ��C�����f�[�^�����m�ł���C�������ɍ쐬���ꂽ���̂ł��邱�Ƃ��m�ۂ����B
�@���̑��ɁC���ۂɁg�M�����̊�h�Ɋ�Â��Ď������s���ꍇ�̗v������߂��Ă���C�u�V���i���̐\�������̐M�����̊�̏���ɂ��āv5�j�ɁC���̂悤�ɋL�ڂ���Ă���B
��@��̏���̐����̐���
�@���i�i�ȉ��u�V���i���v�Ƃ����B�j�̏��F�\���҂́C�V���i���̐\�������ɂ��Ċ�ɂ����W�y�э쐬���s�����߂ɕK�v�ȑg�D�y�ѐl��������ƂƂ��ɁC��ւ̓K�������č�����葱�����܂ސ\�������̎��W�y�э쐬�ɌW��Ɩ��̕W���菇���̍쐬�C�W�E���̋���P�����C������炷�邽�߂ɕK�v�ȑ[�u���u����悤�w�߂邱�ƁB
��@�ӔC�҂̒q�y�я���
�@�\�������̎��W�y�э쐬�ɌW��Ɩ�������ӔC�҂́C�\�������̖`���Ɏ��̒q���тɏ������͋L���y�ѓ����s�������ʂ�Y�t���邱�ƁB
�A�@�\�������́C���������ɏ]���Ď��W���C�쐬�������̂ɑ���Ȃ��|�̒q
�C�@�ӔC�҂̏������͋L���y�ѓ��
�O�@���̑��i�ȗ��j
�@�����ō쐬�����߂Ă���菇���ɂ��āC���҂炪�̗p���Ă��镶���Ǘ��Ɋւ���菇���̍��ڂɂ��āC���̗�������B
�@�����Ǘ��Ɋւ���SOP�̍��ځi��j
�@�@1�D�ړI
�@�@2�D�K�p�͈�
�@�@3�D�ӔC�҂Ɩ���
�@�@3.1 �i���ۏؐӔC�҂̖���
�@�@3.2 �����Ǘ��ӔC�҂̖���
�@�@4�D�Ǘ��̑ΏۂƂȂ镶���y�ыL�^
�@�@4.1 �����̌n
�@�@4.2 �����̕ۊǁE�Ǘ�
�@�@4.3 �����Ǘ��䒠�̍쐬
�@�@5�D�����y�ыL�^�̊Ǘ��菇
�@�@5.1 �쐬�C����
�@�@5.2 ���F
�@�@5.3 �p�~
�@�@5.4 �����ԍ��̕t��
�@�@5.5 �z�z
�@�@5.6 ���
�@�@5.7 �ۊ�
�@�@5.8 �p��
�@�@�ʎ��F�l��
�@�@�ʓY�F�����y�ыL�^�̊Ǘ��Ɋւ���t���[�`���[�g
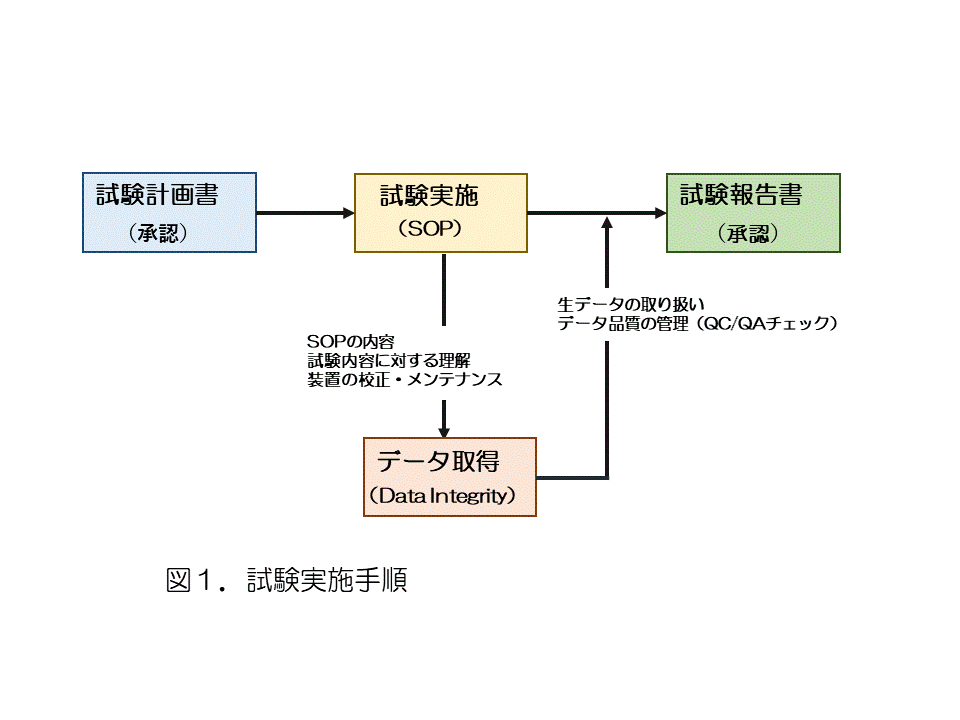
�@�������āC�����̉�Ђł́g�M������h�����炷�邽�߂̊����SOP�𐧒肵�C���������菇���ɏ]���Ď������s���Ă���B���ɁC���ۂɎ������s���C�f�[�^���擾���邽�߂̎菇�ɂ��Đ}1����ɏЉ��6�j�B
�@�@�@ �����v�揑���쐬����i���̍쐬�菇�́CSOP �����Ă����ׂ��ł���j�B
�@�A�@ �����v�揑�̓��e�ɂ��āC�W�҂��ƍ������F���s���B���̎��C�J���i�K�ł́C�K�������i���ۏؕ���i���邢�͒S���ҁj������킯�ł͂Ȃ��B���̂��ߏ��F�͕���̏�i���邢�͑�O�ғI�Ȑl���s�����Ƃł悢�B
�@�B�@ �������s���C���̓��e���L�^����B���̋L�^�ł��邪�C1�����̗p���ł͂Ȃ������m�[�g�̂悤�ɁC�����y�[�W������ꂽ��킩��悤�Ȃ��̂��g�p����B�܂��C�f�[�^��L�^���L�ڂ������t�C�T�C�����L�ڂ���B�����C�`���[�g����t����ꍇ�C�͂���Ȃ��悤�ɂ��邱�ƂƊ�����s���C�ォ�璣��ւ����Ȃ��悤�ɂ���B�������M�����g���Ă���ꍇ�C�R�s�[�����C����l�̎菇�Œ���t���邱�Ƃ��]�܂����B
�@�C�@ ���ׂĂ̎������I��������C�L�^�Ɋ�Â��ĕ����쐬����B���̎��ɁC�]�L�~�X���Ȃ��悤�ɏ\�����ӂ���ƂƂ��ɁC�ł���Α�O�ғI�ȗ���̐l�ɋL�ړ��e�Ƀ~�X�����邩�ǂ������m�F���Ă��炢�C���̏�ŏ��F��B���̎菇�́C�A�̎菇�Ɠ����ł��悢�B
�@�D�@ �������̏��F���̂��C���ׂĂ̋L�^���܂߁C������ׂ��ꏊ�ɕۊNJǗ�����B���������ۊǂ��ꂽ�����v�揑�E���E�L�^��������ꍇ�̎菇�iSOP�j��݂��C�����̕�����h���B�ł���C���������������X�g���쐬���C�ݏo���ɂ��Ċm�F�ł���悤�ɂ��邱�Ƃ��|�C���g�ɂȂ�B
�@�����ł����Ζ��ƂȂ�̂��C��w�Ɍ������ϑ������ꍇ�C���̌��ʂ��g�M�����̊�h�ɓK�����Ă��Ȃ����߁C�\�������Ƃ��Ďg�p�ł��Ȃ������͎�������蒼���ꍇ���������Ă���B�Ⴆ�C���̌��ɊW���Ď��̂悤�ȋ^�₪�N����7�j�B
�@�^��F ���͂𗠕t���鎎���̐M�����m�ۂɂ����āC����������w�Ŏ��{����ꍇ�ł��C�\���p�̃f�[�^�Ƃ���ꍇ�́C��ƂƓ������x���ł̊Ǘ������߂��܂����B��w���ɂǂ��܂ŋ��߂�ׂ��ł����i�@��ނ̍Z���C�g�p�̋L�^�C�����E����̕ۊǂ̋L�^���C�ǂ��܂ł̐��f�[�^�����߂��܂����j�B
�@�F �\���p�̎����f�[�^�Ƃ���ꍇ�C��ƂƓ������x���̊Ǘ��Ƃ���܂����C�\���p�̎����̐M�����m�ۂ̂�����́C�{�݂ɂ��l�X�ŁC�Ȃ��ɂ͉ߏ�ȑΉ������Ă���{�݂�����܂��B�����̐M�����m�ۂ̊�{�́C�������甭�����鐶�f�[�^�E�L�^�ނ��ۑ�����C�������č\���ł��邱�Ƃɂ���܂��B���̗v�������Ă���C�]�������Ƃ��Đ\���Ɏg�p���邱�Ƃ��\�ƍl���܂��B��w�Ŏ��{���鎎���ɂ����Ă��C�����̋L�^�Ƃ��āC����̋L�^�C���ʂ̋L�^�C�Ǘ��̋L�^���������܂����C����ƌ��ʂ̋L�^�͕K�{�ŁC�K�ȊǗ����K�v�ƂȂ�B�Ǘ��̋L�^�Ƃ��āC�@��̏ꍇ�C�K���ɍ쓮����@����g�p�������Ƃ��m�F�ł��邱�Ƃ��K�v�B���̂��߂ɂ́C�������{���̋@��̏��m�F�ł���L�^�i�_����Z���E�C�����j��ۑ����Ă����K�v������B�@��̎g�p�ɂ��ẮC����̋L�^�̒��ɋ@������ł���L�ڂ�����悢�B�����⎎��́C�K�ȕi���̂��̂��g�p�������Ƃ��L�^�i����L�^��i���L�^�j�v����m�F�ł��邱�Ƃ��K�v�B�Ȃ��C�ۊǂ��������⎎����g�p�����ꍇ�C�K���ɕۊǂ����L�^���K�v�B
�@�����������ւ̑Ή��Ƃ��āC���R��w�ł́C���̂悤�Ȏ��g�݂��s���Ă���ƕ���Ă���8�j�B
�@�A�J�f�~�A�݂̂Ő��ݏo���ꂽ����≻�����̗Տ��J���i�K�ł̐������́C�Y�w�A�g�ɔ�חL�ӂɒႢ�B���̗��R�Ƃ��āC���i�J����ړI�Ƃ����Ƃ̉��p�����ƁC�\�z�O�̌��ʂ���ɁC���̌��ۂ��Ȋw�I�ɏؖ������Ր�����������A�J�f�~�A�̊�b�����Ƃ̌����u�����̈Ⴂ���������Ă���B�������ӌ��̒��ɂ́C�A�J�f�~�A�̎����f�[�^�ɂ��āC�Č������Ⴍ�C�M�����Ɍ�����Ƃ̎w�E������B���R��w�ł́C�M�����ɌW��u������J�Â��Ă���C�����ōu�t����̓I�Ɏw�E���������L�^�̖��_���w�E���C������[�߂Ă���B
�@�����������Ƃ���g�M�����̊�h�Ɋւ���F�m�x�̌���C�����đ�w�̏����P����Ă���̂ł͂Ȃ����ƍl���Ă���B�����C�S�z������悤�ł���C�����҂��w�ɔh�����đΉ����铙�̎�i���K�v�ł��낤�B
�Q�l����
1�j�����J���ȁC�@�{�s�K����43���C�����J���ȗߑ�121������17�N7��25��
2�j���a���j�C�M�����ƈ��S���CSEC journal�C10 �i3�j�C8-10 �i2014�j
3�j��R���m��C����F�̎d�g�݂ɂ��� �`���S�Ō��ʓI�Ȗ�܂�͂��邽�߂Ɂ`�CJCCG-TOP2
��������Q�m����Ãt�H�[���� 2023�N1��21��
https://www.pmda.go.jp/files/000263354.pdf
4�j���{���N��Õ��C�����Ƃ̐ӔC�����̕��� �`���҂���̈��S�E���S����邽�߂Ɂ`�@�ߘa5�N3��
https://www.pref.osaka.lg.jp/attach/31416/00272947/sekinin_yakuin.pdf
5�j�����ȁC�V���i���̐\�������̐M�����̊�̏���ɂ��āC���R��1058���C����10�N12��1���C
6�j�{�����t�C�J���i�K�ɉ������o���f�[�V�������{�͈́E�i���K�i�ݒ�ƕύX�Ǘ��|�v���Z�X�^���͖@�o���f�[�V�����|�C�T�C�G���X&�e�N�m���W�[
�i���j �i2023�j
7�j�T�C�G���X&�e�N�m���W�[ �i���j�C�����S���҂�������Y�݂ւ̉I�M������K�p�����ł̎��{��yQ&A�W/SOP��z�i2021�j
16�j�����c���M�C�A�J�f�~�A�ɂ���������L�^�̔Y�� �������������ɂ́C YAKUGAKU
ZASSHI 139�C 887-890 �i2019�j
|



